‘heritability’ directory
- See Also
- Gwern
- Links
- “Population-Specific Polygenic Risk Scores for People of Han Chinese Ancestry”, Chen et al 2025
- “Within-Family Heritability Estimates for Behavioral and Disease Phenotypes from 500,000 Sibling Pairs of Diverse Ancestries”, Yengo et al 2025
- “Direct Effect of Genetic Ancestry on Complex Traits in a Mexican Population”, Wang et al 2025
- “Importance of Clinical, Laboratory, and Genetic Risk Factors for Incident CAD”, Bhattacharya et al 2025
- “Polygenic Score Prediction Within and Between Sibling Pairs for Intelligence, Cognitive Abilities, and Educational Traits From Childhood to Early Adulthood”, Lin et al 2025
- “Three Promising Directions in the Study of Intelligence With Genetic Methods”, Lee & Morris 2025b
- “Nature, Nurture, and Socioeconomic Outcomes: New Evidence from Sib Pairs and Molecular Genetic Data”, Markel et al 2025
- “A Litmus Test for Confounding in Polygenic Scores”, Smith et al 2025
- “Trans-Ancestry Genome-Wide Study of Depression Identifies 697 Associations Implicating Cell Types and Pharmacotherapies”, Consortium 2025
- “Within-Family GWAS Does Not Ameliorate the Decline in Prediction Accuracy across Populations”, Zhang & Conley 2024
- “Genetically-Diverse Crowds Are Wiser”, Barneron et al 2024
- “DNA and IQ: Big Deal or Much Ado about Nothing? A Meta-Analysis”, Oxley et al 2024
- “Dietary Restriction Impacts Health and Lifespan of Genetically Diverse Mice”, Francesco et al 2024
- “Analysis of 3.6 Million Individuals Yields Minimal Evidence of Pairwise Genetic Interactions for Height”, Jabalameli et al 2024
- “Effect of Disclosing a Polygenic Risk Score for Coronary Heart Disease on Adverse Cardiovascular Events: 10-Year Follow-Up of the MI-GENES Randomized Clinical Trial”, Naderian et al 2024
- “Efficient BlockLASSO for Polygenic Scores With Applications to All of Us and UK Biobank”, Raben et al 2024
- “A 30-Nation Investigation of Lay Heritability Beliefs”, Ferris et al 2024
- “Star Trek Fan Leaves Behind a Collection Like No One Has Done Before: When Troy Nelson Died, His Shelves Were Filled to the Rafters With Memorabilia from the Popular Franchise. Soon, the Massive Collection Will Be Boldly Going, Going, Gone”, Deb 2024
- “Predicting the Direction of Phenotypic Difference”, Gokhman et al 2024
- “The Nurture of Nature and the Nature of Nurture: How Genes and Investments Interact in the Formation of Skills”, Houmark et al 2024
- “Gene-Environment Correlation: The Role of Family Environment in Academic Development”, Zhou et al 2024
- “Refining the Impact of Genetic Evidence on Clinical Success”, Minikel et al 2023
- “Genetic Influences on Educational Attainment Through the Lens of the Evolving Swedish Welfare State: A Cross-Level Gene-Environment Interaction Study Based on Polygenic Indices and Longitudinal Register Data”, Pettersson 2023
- “Assessing the Impact of 20th Century Internal Migrations on the Genetic Structure of Estonia”, Kuznetsov et al 2023
- “The Heritability of Ability Tilts”, Coyle et al 2023
- “Multi-Ancestry Polygenic Mechanisms of Type 2 Diabetes Elucidate Disease Processes and Clinical Heterogeneity”, Smith et al 2023
- “It Matters What and Where We Measure: Education and Ideology in a Swedish Twin Design”, Ahlskog 2023
- “A Biobank-Scale Test of Marginal Epistasis Reveals Genome-Wide Signals of Polygenic Epistasis”, Fu et al 2023
- “GWAS of Random Glucose in 476,326 Individuals Provide Insights into Diabetes Pathophysiology, Complications and Treatment Stratification”, Lagou et al 2023
- “Population-Level Genetic Variation Shapes Generative Brain Mechanisms”, Monaghan et al 2023
- “Principles and Methods for Transferring Polygenic Risk Scores across Global Populations”, Kachuri et al 2023
- “Deep Learning Based Phenotyping of Medical Images Improves Power for Gene Discovery of Complex Disease”, Flynn et al 2023
- “Nature and Nurture in Fussy Eating from Toddlerhood to Early Adolescence: Findings from the Gemini Twin Cohort”, Nas et al 2023
- “Overcoming Attenuation Bias in Regressions Using Polygenic Indices”, Kippersluis et al 2023
- “Estimation of Indirect Genetic Effects and Heritability under Assortative Mating”, Young 2023
- “Multi-Ancestry Genome-Wide Meta-Analysis of 56,241 Individuals Identifies LRRC4C, LHX5-AS1 and Nominates Ancestry-Specific Loci PTPRK, GRB14, and KIAA0825 As Novel Risk Loci for Alzheimer Disease: the Alzheimer Disease Genetics Consortium”, Rajabli et al 2023
- “Rapid and Accurate Multi-Phenotype Imputation for Millions of Individuals”, Gu et al 2023
- “The Relationship between Genotype- and Phenotype-Based Estimates of Genetic Liability to Human Psychiatric Disorders, in Practice and in Theory.”, Krebs et al 2023
- “Do Polygenic Indices Capture ‘Direct’ Effects on Child Externalizing Behavior? Within-Family Analyses in Two Longitudinal Birth Cohorts”, Tanksley et al 2023
- “Shaping Faces: Genetic and Epigenetic Control of Craniofacial Morphogenesis”, Selleri & Rijli 2023
- “Gene-Environment Interplay in Early Life Cognitive Development”, Stumm et al 2023
- “The Big (Genetic) Sort? Reassessing Migration Patterns and Their Genetic Imprint in the UK”, Furuya et al 2023
- “Identification and Analysis of Individuals Who Deviate from Their Genetically-Predicted Phenotype”, Hawkes et al 2023
- “A 12-Gene Pharmacogenetic Panel to Prevent Adverse Drug Reactions: an Open-Label, Multicenter, Controlled, Cluster-Randomized Crossover Implementation Study (PREPARE)”, Swen et al 2023
- “Sharing GWAS Summary Statistics Results in More Citations”, Reales & Wallace 2023
- “Phenotype Integration Improves Power and Preserves Specificity in Biobank-Based Genetic Studies of MDD”, Dahl et al 2023
- “Genetic and Environmental Influences on Playing Video Games”, Hassan 2023
- “15 Years of GWAS Discovery: Realizing the Promise”, Abdellaoui et al 2023
- “Childhood Maltreatment and Mental Health Problems: A Systematic Review and Meta-Analysis of Quasi-Experimental Studies”, Baldwin et al 2023
- “The Heritability of Pescetarianism and Vegetarianism”, Wesseldijk et al 2023
- “PATH: Defining Ancestry, Heritability and Plasticity of Cellular Phenotypes in Somatic Evolution”, Schiffman et al 2022
- “PIGEON: Reimagining Gene-Environment Interaction Analysis for Human Complex Traits”, Miao et al 2022
- “A Genetically Informed Registered Report on Adverse Childhood Experiences and Mental Health”, Baldwin et al 2022
- “Polygenic Risk Scores for the Prediction of Cardiometabolic Disease”, O’Sullivan et al 2022
- “Genetic Diversity Fuels Gene Discovery for Tobacco and Alcohol Use”, Saunders et al 2022
- “Genetic Influences on Educational Achievement in Cross-National Perspective”, Baier et al 2022
- “The APOE Locus Is Linked to Decline in General Cognitive Function: 20-Years Follow-Up in the Doetinchem Cohort Study”, Rietman et al 2022
- “Polygenic Prediction of Molecular Traits Using Large-Scale Meta-Analysis Summary Statistics”, Pain et al 2022
- “Parental Transmission and the Importance of the (Noncausal) Effects of Education on Political Engagement: Missing the Forest for the Trees”, Rasmussen et al 2022
- “Familial Concentration of Crime in a Digital Era: Criminal Behavior among Family Members of Cyber Offenders”, Weijer & Moneva 2022
- “Two Pup Vocalization Types Are Genetically and Functionally Separable in Deer Mice”, Jourjine et al 2022
- “Correcting for Volunteer Bias in GWAS Uncovers Novel Genetic Variants and Increases Heritability Estimates”, Alten et al 2022
- “Advances and Applications of Polygenic Scores for Coronary Artery Disease”, Patel & Khera 2022
- “Heritable and Sex-Specific Variation in the Development of Social Behavior in a Wild Primate”, Lange et al 2022
- “Benefits and Barriers to Implementing Precision Preventive Care: Results of a National Physician Survey”, Vassy et al 2022
- “Polygenic Scoring Accuracy Varies across the Genetic Ancestry Continuum in All Human Populations”, Ding et al 2022
- “Sibling Variation in Phenotype and Genotype: Polygenic Trait Distributions and DNA Recombination Mapping With UK Biobank and IVF Family Data”, Lello et al 2022
- “Improving on Polygenic Scores across Complex Traits Using Select and Shrink With Summary Statistics”, Tyrer et al 2022
- “Extremely Sparse Models of Linkage Disequilibrium in Ancestrally Diverse Association Studies”, Nowbandegani et al 2022
- “What Does Heritability of Alzheimer’s Disease Represent?”, Baker et al 2022
- “The Phenotype-Genotype Reference Map: Improving Biobank Data Science through Replication.”, Bastarache et al 2022
- “Global Biobank Analyses Provide Lessons for Developing Polygenic Risk Scores across Diverse Cohorts”, Wang et al 2022
- “Asymmetrical Genetic Attributions for the Presence and Absence of Health Problems”, Lebowitz et al 2022
- “Concordance for Gender Dysphoria in Genetic Female Monozygotic (Identical) Triplets”, Kauffman et al 2022
- “Molecular Genetics and Mid-Career Economic Mobility”, Minard 2022
- “With LeBron James Junior Nearly Grown, Could Dad’s Preposterous NBA Dream Become Reality? LeBron James’s Last NBA Goal Might Be to Play Long Enough to Take the Court With His Oldest Son. But Is Bronny James a Legit NBA Prospect, and What Would It Take for the Lakers to Turn Father and Son into Teammates?”, Sherman 2022
- “Cost-Effectiveness of Polygenic Risk Scores to Guide Statin Therapy for Cardiovascular Disease Prevention”, Kiflen et al 2022
- “A Polygenic Risk Score to Predict Sudden Cardiac Arrest in Patients With Coronary Artery Disease”, Porcu et al 2022
- “Health Care Utilization of Fine-Scale Identity by Descent Clusters in a Los Angeles Biobank”, Caggiano et al 2022
- “From Mendel to Quantitative Genetics in the Genome Era: the Scientific Legacy of W. G. Hill”, Charlesworth et al 2022
- “Lithium Response in Bipolar Disorder: Genetics, Genomics, and Beyond”, Papiol et al 2022
- “Systematic Comparison of Family History and Polygenic Risk across 24 Common Diseases”, Mars et al 2022
- “Medical Genetics in the 19th Century As Background to the Development of Psychiatric Genetics”, Kendler 2022
- “Physical Activity and Health: Findings from Finnish Monozygotic Twin Pairs Discordant for Physical Activity”, Kujala et al 2022
- “Examining Social Genetic Effects on Educational Attainment via Parental Educational Attainment, Income, and Parenting”, Su et al 2022
- “Clarifying the Causes of Consistent and Inconsistent Findings in Genetics”, Dattani et al 2022
- “An Anatomy of the Intergenerational Correlation of Educational Attainment—Learning from the Educational Attainments of Norwegian Twins and Their Children”, Baier et al 2022
- “Sex Differences in the Genetic and Environmental Underpinnings of Meat and Plant Preferences”, Çınar et al 2022
- “Secretoglobin Family 1D Member 2 (SCGB1D2) Protein Inhibits Growth of Borrelia Burgdorferi and Affects Susceptibility to Lyme Disease”, Strausz et al 2022
- “How Full Is the Evolutionary Fuel Tank? A Meta-Analysis Quantifies the Heritable Genetic Variance in Fitness—The Fuel of Evolution”, Walsh 2022
- “Human Brain Anatomy Reflects Separable Genetic and Environmental Components of Socioeconomic Status”, Kweon et al 2022
- “Reweighting the UK Biobank to Reflect Its Underlying Sampling Population Substantially Reduces Pervasive Selection Bias due to Volunteering”, Alten et al 2022
- “Fast and Accurate Bayesian Polygenic Risk Modeling With Variational Inference”, Zabad et al 2022
- “Evaluating Indirect Genetic Effects of Siblings Using Singletons”, Howe et al 2022
- “Birth Order Differences in Education Originate in Postnatal Environments”, Isungset et al 2022
- “Epistasis and Adaptation on Fitness Landscapes”, Bank 2022
- “Genomics, Convergent Neuroscience and Progress in Understanding Autism Spectrum Disorder”, Willsey et al 2022
- “Multi-Omics Analyses Cannot Identify True-Positive Novel Associations from Underpowered Genome-Wide Association Studies of Four Brain-Related Traits”, Baranger et al 2022
- “Childhood Trauma and Borderline Personality Disorder Traits: A Discordant Twin Study”, Skaug et al 2022
- “Dominance versus Epistasis: the Biophysical Origins and Plasticity of Genetic Interactions within and between Alleles”, Xie et al 2022
- “Genetic and Environmental Influences on Biological Essentialism, Heuristic Thinking, Need for Closure, and Conservative Values: Insights From a Survey and Twin Study”, Morosoli et al 2022
- “New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias”, Bellenguez et al 2022
- “Generalizability and Effect Measure Modification in Sibling Comparison Studies”, Sjölander et al 2022
- “Sibling Comparison Studies”, Sjölander et al 2022
- “Modeling Assortative Mating and Genetic Similarities between Partners, Siblings, and In-Laws”, Torvik et al 2022
- “Multivariate Genomic Architecture of Cortical Thickness and Surface Area at Multiple Levels of Analysis”, Grotzinger et al 2022
- “Quantifying Bias from Measurable & Unmeasurable Confounders Across 3 Domains of Individual Determinants of Political Preferences”, Ahlskog & Oskarsson 2022
- “Significant Sparse Polygenic Risk Scores across 813 Traits in UK Biobank”, Tanigawa et al 2022
- “Patterns of Item Nonresponse Behavior to Survey Questionnaires Are Systematic and Have a Genetic Basis”, Mignogna et al 2022
- “The Genetics of Specific Cognitive Abilities”, Procopio et al 2022
- “Measuring Biodiversity from DNA in the Air”, Clare et al 2022
- “Discovery of Genomic Loci of the Human Cerebral Cortex Using Genetically Informed Brain Atlases”, Makowski et al 2022
- “Idiosyncratic Learning Performance in Flies”, Smith et al 2022
- “Multi-Ancestry EQTL Meta-Analysis of Human Brain Identifies Candidate Causal Variants for Brain-Related Traits”, Zeng et al 2022
- “Alcohol Metabolism Genes and Risks of Site-Specific Cancers in Chinese Adults: An 11-Year Prospective Study”, Im et al 2022
- “ExPRSweb—An Online Repository With Polygenic Risk Scores for Common Health-Related Exposures”, Ma et al 2022
- “Interaction Testing and Polygenic Risk Scoring to Estimate the Association of Common Genetic Variants With Treatment Resistance in Schizophrenia”, Pardiñas et al 2022
- “A Saturated Map of Common Genetic Variants Associated With Human Height from 5.4 Million Individuals of Diverse Ancestries”, Yengo et al 2022
- “Identifying Imaging Genetic Associations via Regional Morphometricity Estimation”, Bao et al 2022
- “Familial Resemblance, Citizenship, and Counterproductive Work Behavior: A Combined Twin, Adoption, Parent-Offspring, and Spouse Approach”, Anderson et al 2022b
- “Role of Polygenic Risk Score in the Familial Transmission of Bipolar Disorder in Youth”, Birmaher et al 2021
- “Interest of Phenomic Prediction As an Alternative to Genomic Prediction in Grapevine”, Brault et al 2021
- “Evidence for Excess Familial Clustering of Post Traumatic Stress Disorder in the US Veterans Genealogy Resource”, Cannon-Albright et al 2021
- “Multi-Ancestry Meta-Analysis of Asthma Identifies Novel Associations and Highlights the Value of Increased Power and Diversity”, Tsuo et al 2021
- “Dietary Mediators of the Genetic Susceptibility to Obesity?Results from the Quebec Family Study”, Jacob et al 2021
- “Global Biobank Meta-Analysis Initiative: Powering Genetic Discovery across Human Diseases”, Initiative & Zhou 2021
- “The Genetic Architecture of Obsessive-Compulsive Disorder: Contribution of Liability to OCD From Alleles Across the Frequency Spectrum”, Mahjani et al 2021
- “Genetically Informed, Multilevel Analysis of the Flynn Effect across 4 Decades & 3 WISC Versions”, Giangrande et al 2021
- “Improving GWAS Discovery and Genomic Prediction Accuracy in Biobank Data”, Orliac et al 2021
- “Returning Actionable Genomic Results in a Research Biobank: Analytic Validity, Clinical Implementation, and Resource Usage”, Zawatsky et al 2021
- “Bridging the Explanatory Gaps: What Can We Learn from a Biological Agency Perspective?”, Sultan et al 2021
- “Parent-Of-Origin Effects in the UK Biobank”, Hofmeister et al 2021
- “Genome-Wide Analysis of 53,400 People With Irritable Bowel Syndrome Highlights Shared Genetic Pathways With Mood and Anxiety Disorders”, Eijsbouts et al 2021
- “Sex Impacts the Function of Major Depression-Linked Variants in Vivo”, Mulvey et al 2021
- “The Genetic Basis of Spatial Cognitive Variation in a Food-Caching Bird”, Branch et al 2021
- “Predicting Skeletal Stature Using Ancient DNA”, Cox et al 2021
- “Between-Group Mean Differences in Intelligence in the United States Are >0% Genetically Caused: Five Converging Lines of Evidence”, Warne 2021b
- “Genomic Prediction in the Wild: A Case Study in Soay Sheep”, Ashraf et al 2021
- “Polygenic Basis and Biomedical Consequences of Telomere Length Variation”, Codd et al 2021
- “Evolution of Sociability by Artificial Selection”, Scott et al 2021
- “A Gene-Environment Interaction Study of Polygenic Scores and Maltreatment on Childhood ADHD”, He & Li 2021b
- “Increased Somatic Mutation Burdens in Normal Human Cells due to Defective DNA Polymerases”, Robinson et al 2021
- “Investigating Perceived Heritability of Mental Health Disorders and Attitudes toward Genetic Testing in the United States, United Kingdom, and Australia”, Morosoli et al 2021
- “Pervasive Downward Bias in Estimates of Liability Scale Heritability in GWAS Meta-Analysis: A Simple Solution”, Grotzinger et al 2021
- “The Debate Between Two of the Founders of American Psychiatric Genetics, Aaron Rosanoff and Abraham Myerson, on Mendelian Models for Psychiatric Illness (1911–1917)”, Kendler 2021b
- “The Association between Polygenic Scores for Attention-Deficit/hyperactivity Disorder and School Performance: The Role of Attention-Deficit/hyperactivity Disorder Symptoms, Polygenic Scores for Educational Attainment, and Shared Familial Factors”, Jangmo et al 2021
- “Genome-Wide Association Study of Musical Beat Synchronization Demonstrates High Polygenicity”, Niarchou et al 2021
- “Using Genes to Explore the Effects of Cognitive and Non-Cognitive Skills on Education and Labor Market Outcomes”, Buser et al 2021 (page 3)
- “Discovery of 42 Genome-Wide Statistically-Significant Loci Associated With Dyslexia”, Doust et al 2021
- “Leveraging Fine-Mapping and Non-European Training Data to Improve Cross-Population Polygenic Risk Scores”, Weissbrod et al 2021
- “Somatic Mutation Rates Scale With Lifespan across Mammals”, Cagan et al 2021
- “The Omnigenic Model and Polygenic Prediction of Complex Traits”, Mathieson 2021
- “The Distribution of Common-Variant Effect Sizes”, O’Connor 2021
- “Genome-Wide Methylation Data Improves Dissection of the Effect of Smoking on Body Mass Index”, Amador et al 2021
- “MegaLMM: Mega-Scale Linear Mixed Models for Genomic Predictions With Thousands of Traits”, Runcie et al 2021
- “X-Chromosome Influences on Neuroanatomical Variation in Humans”, Mallard et al 2021b
- “The Nature of Hereditary Influences on Insanity from Research on Asylum Records in Western Europe in the Mid-19th Century”, Kendler 2021
- “Genome Scans of Facial Features in East Africans and Cross-Population Comparisons Reveal Novel Associations”, Liu et al 2021
- “Genetics of Substance Use Disorders in the Era of Big Data”, Gelernter & Polimanti 2021
- “The Chilean Socio-Ethno-Genomic Cline”, Barozet et al 2021
- “Blood-Based Epigenome-Wide Analyses of Cognitive Abilities”, McCartney et al 2021
- “Rapid Sequencing-Based Diagnosis of Thiamine Metabolism Dysfunction Syndrome”, Owen et al 2021
- “Genomic Characterization of World’s Longest Selection Experiment in Mouse Reveals the Complexity of Polygenic Traits”, Palma-Vera et al 2021
- “Bi-Ancestral Depression GWAS in the Million Veteran Program and Meta-Analysis in >1.2 Million Individuals Highlight New Therapeutic Directions”, Levey et al 2021
- “Loss-Of-Function Mutations in the Melanocortin 4 Receptor in a UK Birth Cohort”, Wade et al 2021
- “Genome-Wide Association Study of More Than 40,000 Bipolar Disorder Cases Provides New Insights into the Underlying Biology”, Mullins et al 2021
- “Echolocating Bats Rely on an Innate Speed-Of-Sound Reference”, Amichai & Yovel 2021
- “Resource Profile and User Guide of the Polygenic Index Repository”, Becker et al 2021
- “Multi-Scale Inference of Genetic Trait Architecture Using Biologically Annotated Neural Networks”, Demetci et al 2021
- “A Comparison of Ten Polygenic Score Methods for Psychiatric Disorders Applied across Multiple Cohorts”, Ni et al 2021
- “Gene-Environment Correlations and Causal Effects of Childhood Maltreatment on Physical and Mental Health: a Genetically Informed Approach”, Warrier et al 2021
- “Modification of Heritability for Educational Attainment and Fluid Intelligence by Socioeconomic Deprivation in the UK Biobank”, Rask-Andersen et al 2021
- “Genetic and Environmental Sources of Familial Resemblance in Anxiety: a Nuclear Twin Family Design”, Ding et al 2021
- “An Expanded Set of Genome-Wide Association Studies of Brain Imaging Phenotypes in UK Biobank”, Smith et al 2021c
- “Are These Australia’s Most Identical Twins? Meet the Twinnies: Bridgette and Paula Powers”, News 2021
- “Toward a Fine-Scale Population Health Monitoring System”, Belbin et al 2021
- “Genetic Attributions and Perceptions of Naturalness Are Shaped by Evaluative Valence”, Lebowitz et al 2021
- “Parental Characteristics and Offspring Mental Health and Related Outcomes: a Systematic Review of Genetically-Informative Literature”, Jami et al 2021
- “Large Uncertainty in Individual PRS Estimation Impacts PRS-Based Risk Stratification”, Ding et al 2021
- “Polygenic Risk for Depression, Anxiety and Neuroticism Are Associated With the Severity and Rate of Change in Depressive Symptoms across Adolescence”, Kwong et al 2021
- “Evaluation of Polygenic Prediction Methodology within a Reference-Standardized Framework”, Pain et al 2021
- “High Trait Variability in Optimal Polygenic Prediction Strategy within Multiple-Ancestry Cohorts”, Lehmann et al 2021
- “Using DNA to Predict Intelligence”, Stumm & Plomin 2021
- “GWAS in Almost 195,000 Individuals Identifies 50 Previously Unidentified Genetic Loci for Eye Color”, Simcoe et al 2021
- “Genes, Ideology, and Sophistication”, Kalmoe & Johnson 2021
- “Therapygenetic Effects of 5-HTTLPR on Cognitive-Behavioral Therapy in Anxiety Disorders: A Meta-Analysis”, Schiele et al 2021
- “Using DNA to Predict Behavior Problems from Preschool to Adulthood”, Gidziela et al 2021
- “Phenotypic Covariance across the Entire Spectrum of Relatedness for 86 Billion Pairs of Individuals”, Kemper et al 2021
- “The Augmented Classical Twin Design: Incorporating Genome-Wide Identity by Descent Sharing Into Twin Studies in Order to Model Violations of the Equal Environments Assumption”, Hwang et al 2021
- “Pathfinder: A Gamified Measure to Integrate General Cognitive Ability into the Biological, Medical and Behavioral Sciences”, Malanchini et al 2021
- “Identification of 370 Genetic Loci for Age at First Sex and Birth Linked to Externalizing Behavior”, Mills et al 2021
- “The Heritability of Reading and Reading-Related Neurocognitive Components: A Multi-Level Meta-Analysis”, Andreola et al 2021
- “Genetics and Child Development: Recent Advances and Their Implications for Developmental Research”, Armstrong-Carter et al 2021
- “Genetic and Environmental Influences of Dietary Indices in a UK Female Twin Cohort”, Mompeo et al 2021
- “Developmental Trajectories of Delinquent and Aggressive Behavior: Evidence for Differential Heritability”, Isen et al 2021
- “From Genotype to Phenotype: Polygenic Prediction of Complex Human Traits”, Raben et al 2021
- “Nurture Might Be Nature: Cautionary Tales and Proposed Solutions”, Hart et al 2021
- “Differences between Germline Genomes of Monozygotic Twins”, Jonsson et al 2021
- “Twin-Singleton Comparisons Across Multiple Domains of Life”, Willemsen et al 2021
- “Dr Rona Strawbridge”
- “The Genetic Architecture of Depression in Individuals of East Asian Ancestry: A Genome-Wide Association Study”, Giannakopoulou et al 2021
- “Evidence of Horizontal Indirect Genetic Effects in Humans”, Xia et al 2020
- “Does the Brain-Derived Neurotrophic Factor Val66Met Polymorphism Modulate the Effects of Physical Activity and Exercise on Cognition?”, Heras et al 2020
- “10 Years of Enhancing Neuro-Imaging Genetics through Meta-Analysis: An Overview from the ENIGMA Genetics Working Group”, Medland et al 2020
- “Largest GWAS (n = 1,126,563) of Alzheimer’s Disease Implicates Microglia and Immune Cells”, Wightman et al 2020
- “The Heritability of Insomnia: A Meta-Analysis of Twin Studies”, Barclay et al 2020
- “Twins Living Apart: Behavioral Insights/Twin Study Reviews: Managing Monochorionic-Diamniotic Twin Pregnancies; Paternity Testing in Multiple Pregnancies; Twin Research on Resilience; Trisomies in Twin Pregnancies/Human Interest: Reunited Brazilian Twins; Website for Twins With Disabled Co-Twins; Twins Separated in Secret of the Nile Series; Mengele: Unmasking the Angel of Death; Twins Helping Others”, Segal 2020
- “An Integrative Analysis of Genomic and Exposomic Data for Complex Traits and Phenotypic Prediction”, Zhou & Lee 2020
- “Estimation of Non-Additive Genetic Variance in Human Complex Traits from a Large Sample of Unrelated Individuals”, Hivert et al 2020
- “‘Landmark’ Study Resolves a Major Mystery of How Genes Govern Human Height”, Kaiser 2020
- “Individuals With Common Diseases but With a Low Polygenic Risk Score Could Be Prioritized for Rare Variant Screening”, Lu et al 2020c
- “Could Polygenic Risk Scores Be Useful in Psychiatry? A Review”, Murray et al 2020
- “Exploring the Variance in Complex Traits Captured by DNA Methylation Assays”, Battram et al 2020
- “Happiness and Wellbeing; the Value and Findings from Genetic Studies”, Weijer et al 2020
- “Genome-Wide Association Study of over 40,000 Bipolar Disorder Cases Provides Novel Biological Insights”, Mullins et al 2020
- “Causes of Variation in Food Preference in the Netherlands”, Vink et al 2020
- “Genetic Fortune: Winning or Losing Education, Income, and Health”, Kweon et al 2020 (page 4)
- “Improved Genetic Prediction of Complex Traits from Individual-Level Data or Summary Statistics”, Zhang et al 2020
- “Sibling Validation of Polygenic Risk Scores and Complex Trait Prediction”, Lello et al 2020
- “On the Genetic Basis of Political Orientation”, Daws & Weinschenk 2020
- “Genetic and Environmental Variation in Educational Attainment: an Individual-Based Analysis of 28 Twin Cohorts”, Silventoinen et al 2020
- “The Genes We Inherit and Those We Don’t: Maternal Genetic Nurture and Child BMI Trajectories”, Tubbs et al 2020
- “Association of Parental Substance Misuse With Offspring Substance Misuse and Criminality: a Genetically Informed Register-Based Study”, Latvala et al 2020
- “Recent Common Origin, Reduced Population Size, and Marked Admixture Have Shaped European Roma Genomes”, Bianco et al 2020
- “Germline Mutation Rates in Young Adults Predict Longevity and Reproductive Lifespan”, Cawthon et al 2020
- “Efficient Polygenic Risk Scores for Biobank Scale Data by Exploiting Phenotypes from Inferred Relatives”, Truong et al 2020
- “Combined Utility of 25 Disease and Risk Factor Polygenic Risk Scores for Stratifying Risk of All-Cause Mortality”, Meisner et al 2020
- “Discovery of 318 New Risk Loci for Type 2 Diabetes and Related Vascular Outcomes among 1.4 Million Participants in a Multi-Ancestry Meta-Analysis”, Vujkovic et al 2020
- “Preimplantation Genetic Testing for Polygenic Disease Relative Risk Reduction: Evaluation of Genomic Index Performance in 11,883 Adult Sibling Pairs”, Treff et al 2020
- “Human Postprandial Responses to Food and Potential for Precision Nutrition”, Berry et al 2020
- “The Polygenic Architecture of Schizophrenia—Rethinking Pathogenesis and Nosology”, Smeland et al 2020
- “Risk in Relatives, Heritability, SNP-Based Heritability, and Genetic Correlations in Psychiatric Disorders: A Review”, Baselmans et al 2020
- “Large-Scale Genome-Wide Association Study in a Japanese Population Identifies Novel Susceptibility Loci across Different Diseases”, Ishigaki et al 2020
- “The Earliest Origins of Genetic Nurture: The Prenatal Environment Mediates the Association Between Maternal Genetics and Child Development”, Armstrong-Carter et al 2020
- “A Fast and Scalable Framework for Large-Scale and Ultrahigh-Dimensional Sparse Regression With Application to the UK Biobank”, Qian et al 2020
- “Exploring Genetic Contributions to News Use Motives and Frequency of News Consumption: A Study of Identical and Fraternal Twins”, York & Haridakis 2020
- “Genetic Risk Scores for Cardiometabolic Traits in Sub-Saharan African Populations”, Ekoru et al 2020
- “Genome-Wide Analysis Identifies Genetic Effects on Reproductive Success and Ongoing Natural Selection at the FADS Locus”, Mathieson et al 2020
- “GWAS of Depression Phenotypes in the Million Veteran Program and Meta-Analysis in More Than 1.2 Million Participants Yields 178 Independent Risk Loci”, Levey et al 2020
- “Musings on Visscher Et Al 2006”, Visscher 2020
- “Insights into the Genetic Architecture of the Human Face”, White et al 2020
- “Heritability of Affectionate Communication: A Twins Study”, Floyd et al 2020
- “Using Genetics for Social Science”, Harden & Koellinger 2020
- “Sociology, Genetics, and the Coming of Age of Sociogenomics”, Mills & Tropf 2020
- “Beyond Culture and the Family: Evidence from Twin Studies on the Genetic and Environmental Contribution to Values”, Twito & Knafo-Noam 2020
- “Greater Variability in Chimpanzee (Pan Troglodytes) Brain Structure among Males”, DeCasien et al 2020
- “Identifying Genetic Variants Underlying Phenotypic Variation in Plants without Complete Genomes”, Voichek & Weigel 2020
- “Birmingham and Beyond”, Eaves 2020
- “Nicholas G. Martin and the Extended Twin Model”, Maes 2020b
- “The SNP-Based Heritability—A Commentary on Yang Et Al 2010”, Yang 2020
- “Genetic Endowments and Wealth Inequality”, Barth et al 2020
- “The Barbarians Are at the Gate!”, Hatemi 2020
- “Sociopolitical Attitudes Through the Lens of Behavioral Genetics: Contributions from Dr Nicholas Martin”, Verhulst 2020
- “Functional Connectome-Wide Associations of Schizophrenia Polygenic Risk”, Cao et al 2020
- “The Multiplex Model of the Genetics of Alzheimer’s Disease”, Sims et al 2020
- “The Behavioral, Cellular and Immune Mediators of HIV-1 Acquisition: New Insights from Population Genetics”, Powell et al 2020
- “Analysis of Variance When Both Input and Output Sets Are High-Dimensional”, Campos et al 2020
- “The Association of Oxytocin Receptor Gene (OXTR) Polymorphisms Antisocial Behavior: A Meta-Analysis”, Poore & Waldman 2020
- “Genetic Architecture of Complex Traits and Disease Risk Predictors”, Yong et al 2020
- “Using Human Genetics to Understand the Disease Impacts of Testosterone in Men and Women”, Ruth et al 2020
- “Studies of Human Twins Reveal Genetic Variation That Affects Dietary Fat Perception”, Lin et al 2020
- “Theoretical and Empirical Quantification of the Accuracy of Polygenic Scores in Ancestry Divergent Populations”, Wang et al 2020
- “Educational Attainment Polygenic Scores in Hungary: Evidence for Validity and a Historical Gene-Environment Interaction”, Ujma et al 2020
- “Reproducible Genetic Risk Loci for Anxiety: Results From ∼200,000 Participants in the Million Veteran Program”, Levey et al 2020
- “Heritability in Friendship Networks”, Neugart & Yildirim 2020
- “A Brief History of Human Disease Genetics”, Claussnitzer et al 2020
- “Multitrait Analysis of Glaucoma Identifies New Risk Loci and Enables Polygenic Prediction of Disease Susceptibility and Progression”, Craig 2020
- “GWAS of 165,084 Japanese Individuals Identified 9 Loci Associated With Dietary Habits”, Matoba 2020
- “Introducing M-GCTA a Software Package to Estimate Maternal (Or Paternal) Genetic Effects on Offspring Phenotypes”, Qiao 2020
- “Obesity and Eating Behavior from the Perspective of Twin and Genetic Research”, Silventoinen 2020
- “How Genetic and Environmental Variance in Personality Traits Shift across the Life Span: Evidence from a Cross-National Twin Study”, Kandler et al 2020
- “Genome-Wide Analysis Identifies Molecular Systems and 149 Genetic Loci Associated With Income”, Hill et al 2019
- “Beyond SNP Heritability: Polygenicity and Discoverability of Phenotypes Estimated With a Univariate Gaussian Mixture Model”, Holland et al 2019
- “Multivariable G-E Interplay in the Prediction of Educational Achievement”, Allegrini et al 2019
- “The Louisville Twin Study: Past, Present and Future”, Davis et al 2019
- “Genome-Wide Association Studies in Schizophrenia: Recent Advances, Challenges and Future Perspective”, Dennison et al 2019
- “Predicting Educational Achievement from Genomic Measures and Socioeconomic Status”, Stumm et al 2019
- “Genes, Gender Inequality, and Educational Attainment”, Herd et al 2019
- “Population-Specific and Transethnic Genome-Wide Analyses Reveal Distinct and Shared Genetic Risks of Coronary Artery Disease”, Koyama et al 2019
- “Structural Variation in the Sequencing Era”, Ho et al 2019
- “The Propensity for Aggressive Behavior and Lifetime Incarceration Risk: A Test for Gene-Environment Interaction (G × E) Using Whole-Genome Data”, Barnes et al 2019
- “Uganda Genome Resource Enables Insights into Population History and Genomic Discovery in Africa”, Gurdasani et al 2019b
- “Associations of Autozygosity With a Broad Range of Human Phenotypes”, Clark et al 2019
- “Sibling Comparisons Elucidate the Associations between Educational Attainment Polygenic Scores and Alcohol, Nicotine and Cannabis”, Salvatore et al 2019
- “Genome-Wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations”, Peterson et al 2019
- “Genomics of Human Aggression: Current State of Genome-Wide Studies and an Automated Systematic Review Tool”, Odintsova et al 2019
- “Polygenic Score Analysis Of Educational Achievement And Intergenerational Mobility”, Rustichini et al 2019
- “Genetic ‘General Intelligence’, Objectively Determined and Measured”, Fuente et al 2019
- “Software As a Service for the Genomic Prediction of Complex Diseases”, Bolli et al 2019
- “In-Field Whole Plant Maize Architecture Characterized by Latent Space Phenotyping”, Gage et al 2019
- “Children of the Twins Early Development Study (CoTEDS): A Children-Of-Twins Study”, Ahmadzadeh et al 2019
- “The Study of Personality Architecture and Dynamics (SPeADy): A Longitudinal and Extended Twin Family Study”, Kandler et al 2019c
- “Mendelian Gene Discovery: Fast and Furious With No End in Sight”, Bamshad et al 2019
- “Dumb or Smart Asses? Donkey’s (Equus Asinus) Cognitive Capabilities Share the Heritability and Variation Patterns of Human’s (Homo Sapiens) Cognitive Capabilities”, González et al 2019 (page 2)
- “A Prospective Analysis of Genetic Variants Associated With Human Lifespan”, Wright et al 2019
- “The Higher Power of Religiosity Over Personality on Political Ideology”, Ksiazkiewicz & Friesen 2019
- “Leveraging European Infrastructures to Access 1 Million Human Genomes by 2022”, Saunders et al 2019
- “Polygenic and Clinical Risk Scores and Their Impact on Age at Onset of Cardiometabolic Diseases and Common Cancers”, Mars et al 2019
- “Genetic Architecture of Socioeconomic Outcomes: Educational Attainment, Occupational Status, and Wealth”, Liu 2019b
- “A Mother’s Legacy: the Strength of Maternal Effects in Animal Populations”, Moore et al 2019
- “An AI Expert’s Toughest Project: Writing Code to save His Son’s Life”
- “The Chinese National Twin Registry: a ‘Gold Mine’ for Scientific Research”, Gao et al 2019
- “Genomic Risk Score Offers Predictive Performance Comparable to Clinical Risk Factors for Ischaemic Stroke”, Abraham et al 2019
- “Inferring the Nature of Missing Heritability in Human Traits Using Data from the GWAS Catalog”, López-Cortegano & Caballero 2019
- “Unravelling the Genetic Basis of Schizophrenia and Bipolar Disorder With GWAS: A Systematic Review”, Prata et al 2019
- “Gene Regulation and the Architecture of Complex Human Traits in the Genomics Era”, Boutwell & White 2019
- “Genetic Influences on Antisocial Behavior: Recent Advances and Future Directions”, Gard et al 2019
- “Making the Most of Clumping and Thresholding for Polygenic Scores”, Privé et al 2019
- “Breed Differences of Heritable Behavior Traits in Cats”, Salonen et al 2019
- “Heritability of Lifetime Earnings”, Hyytinen et al 2019
- “Genetic Variation across the Human Olfactory Receptor Repertoire Alters Odor Perception”, Trimmer et al 2019
- “Amid Animal Cruelty Debate, 80% of South Korea’s Sniffer Dogs Are Cloned”, Tribune 2019
- “Supplement to No Support for Historic Candidate Gene or Candidate Gene-By-Interaction Hypotheses for Major Depression across Multiple Large Samples”, Border et al 2019
- “Maternal and Fetal Genetic Effects on Birth Weight and Their Relevance to Cardio-Metabolic Risk Factors”, Warrington et al 2019
- “The Heritability of Self-Control: A Meta-Analysis”, Willems et al 2019
- “Is Population Structure in the Genetic Biobank Era Irrelevant, a Challenge, or an Opportunity?”, Lawson et al 2019
- “Is Apostasy Heritable? A Behavior Genetics Study”, Freeman 2019
- “Latent Space Phenotyping: Automatic Image-Based Phenotyping for Treatment Studies”, Ubbens et al 2019
- “A Major Role for Common Genetic Variation in Anxiety Disorders”, Purves et al 2019
- “Simulation of Model Overfit in Variance Explained With Genetic Data”, Derringer 2019
- “Comparing Within-Family and Between-Family Polygenic Score Prediction”, Selzam et al 2019
- “Uncovering the Genetic Architecture of Major Depression”, McIntosh et al 2019
- “Ant Collective Behavior Is Heritable and Shaped by Selection”, Walsh et al 2019
- “Evidence That the Association of Childhood Trauma With Psychosis and Related Psychopathology Is Not Explained by Gene-Environment Correlation: A Monozygotic Twin Differences Approach”, Lecei et al 2019
- “Molecular Support for Heterogonesis Resulting in Sesquizygotic Twinning”, Gabbett et al 2019
- “Fast and Flexible Linear Mixed Models for Genome-Wide Genetics”, Runcie & Crawford 2019
- “Repurposing Large Health Insurance Claims Data to Estimate Genetic and Environmental Contributions in 560 Phenotypes”, Lakhani et al 2019
- “Genome-Wide Meta-Analysis of Depression Identifies 102 Independent Variants and Highlights the Importance of the Prefrontal Brain Regions”, Howard et al 2019
- “Adaptive Phenotypic Plasticity for Life-History and Less Fitness-Related Traits”, Acasuso-Rivero et al 2019
- “Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort”, Bai et al 2019
- “Polygenic Scores: a Public Health Hazard?”, Baverstock 2019
- “Mitochondrial Alterations May Underlie Race-Specific Differences in Cancer Risk and Outcome”, Beebe-Dimmer & Cooney 2019
- “Testing for Family Influences on Obesity: The Role of Genetic Nurture”, Cawley et al 2019
- “Family Networks versus Genetics in Social Outcomes, England 1750–2019 [Slides]”, Clark 2019
- “The Causes and Consequences of Genetic Interactions (Epistasis)”, Domingo et al 2019
- “How Genome-Wide Association Studies (GWAS) Made Traditional Candidate Gene Studies Obsolete”, Duncan et al 2019
- “Genetic Variation, Comparative Genomics, and the Diagnosis of Disease”, Eichler 2019
- “Identifying Facial Phenotypes of Genetic Disorders Using Deep Learning”, Gurovich et al 2019
- “Genetic and Phenotypic Landscape of the Major Histocompatibilty Complex Region in the Japanese Population”, Hirata et al 2019
- “New Insight into Human Sweet Taste: a Genome-Wide Association Study of the Perception and Intake of Sweet Substances”, Hwang et al 2019
- “Comment on ‘Polygenic Scores: A Public Health Hazard?’”, Joyner 2019b
- “Native American Groups Wary of Big U.S. Biobank: Tribes Say Health Officials Were Slow to Consult Them”
- “Towards Clinical Utility of Polygenic Risk Scores”, Lambert et al 2019
- “The Genetics of Human Skin and Hair Pigmentation”, Pavan & Sturm 2019
- “Absolute and Relative Estimates of Genetic and Environmental Variance in Brain Structure Volumes”, Strike et al 2019
- “Benefits and Limitations of Genome-Wide Association Studies”, Tam et al 2019
- “Correlations between Relatives: From Mendelian Theory to Complete Genome Sequence”, Thompson 2019
- “Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine”, Udler et al 2019
- “From R. A. Fisher's 1918 Paper to GWAS a Century Later”, Visscher & Goddard 2019
- “Free Will, Determinism, and Intuitive Judgments About the Heritability of Behavior”, Willoughby et al 2019b
- “Complex Trait Prediction from Genome Data: Contrasting EBV in Livestock to PRS in Humans”, Wray et al 2019
- “The Dynamic Associations Between Cortical Thickness and General Intelligence Are Genetically Mediated”, Schmitt et al 2019
- “Phenotypic Annotation: Using Polygenic Scores to Translate Discoveries From Genome-Wide Association Studies From the Top Down”, Belsky & Harden 2019
- “Viewing Education Policy through a Genetic Lens”, Asbury & Wai 2019
- “No Support for Historical Candidate Gene or Candidate Gene-By-Interaction Hypotheses for Major Depression Across Multiple Large Samples”, Border et al 2019
- “Extreme Morning Chronotypes Are Often Familial and Not Exceedingly Rare: the Estimated Prevalence of Advanced Sleep Phase, Familial Advanced Sleep Phase, and Advanced Sleep-Wake Phase Disorder in a Sleep Clinic Population”, Curtis et al 2019
- “The Nature and Nurture of HEXACO Personality Trait Differences: An Extended Twin Family Study”, Kandler et al 2019b
- “A Genome-Wide Association Study of Bitter and Sweet Beverage Consumption”, Zhong et al 2019
- “Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis”, Sharp et al 2019
- “Asymmetrical Genetic Attributions for Prosocial versus Antisocial Behavior”, Lebowitz et al 2019
- “Genomic Prediction of Complex Disease Risk”, Lello et al 2018
- “Variation in the Heritability of Child Body Mass Index by Obesogenic Home Environment”, Schrempft et al 2018
- “Genetic Consequences of Social Stratification in Great Britain”, Abdellaoui et al 2018
- “Existence and Implications of Population Variance Structure”, Musharoff et al 2018
- “Evidence for Bias of Genetic Ancestry in Resting State Functional MRI”, Altmann & Mourao-Miranda 2018
- “A Twin Study on the Correlates of Voluntary Exercise Behavior in Adolescence”, Schutte et al 2018
- “Polygenicity of Complex Traits Is Explained by Negative Selection”, O’Connor et al 2018
- “Genomic Prediction of Cognitive Traits in Childhood and Adolescence”, Allegrini et al 2018
- “Meta-Analysis of Genome-Wide Association Studies for Body Fat Distribution in 694,649 Individuals of European Ancestry”, Pulit et al 2018
- “Evidence for Gene-Environment Correlation in Child Feeding: Links between Common Genetic Variation for BMI in Children and Parental Feeding Practices”, Selzam et al 2018
- “Genes, Education, and Labor Market Outcomes: Evidence from the Health and Retirement Study”, Papageorge & Thom 2018
- “Genome-Wide Statistically-Significant Regions in 43 Utah High-Risk Families Implicate Multiple Genes Involved in Risk for Completed Suicide”, Coon et al 2018
- “The Genetics of the Mood Disorder Spectrum: Genome-Wide Association Analyses of over 185,000 Cases and 439,000 Controls”, Coleman et al 2018
- “On the Genetic and Environmental Sources of Social and Political Participation in Adolescence and Early Adulthood”, Kornadt et al 2018
- “Modeling Functional Enrichment Improves Polygenic Prediction Accuracy in UK Biobank and 23andMe Data Sets”, Márquez-Luna et al 2018
- “Genomic Underpinnings of Lifespan Allow Prediction and Reveal Basis in Modern Risks”, Timmers et al 2018
- “Multi-Ethnic Genome-Wide Association Study for Atrial Fibrillation”, Roselli et al 2018
- “Study of 300,486 Individuals Identifies 148 Independent Genetic Loci Influencing General Cognitive Function”, Davies et al 2018
- “German Law Allows Use of DNA to Predict Suspects' Looks”, Vogel 2018
- “The Genetic Architecture of Hair Color in the UK Population”, Morgan et al 2018
- “A Multimethodological Study of Preschoolers’ Preferences for Aggressive Television and Video Games”, Jamnik & DiLalla 2018
- “Genome-Wide Association Study of Social Genetic Effects on 170 Phenotypes in Laboratory Mice”, Baud et al 2018
- “On the Genetic and Genomic Basis of Aggression, Violence, and Antisocial Behavior”, Beaver et al 2018
- “Novel Susceptibility Loci and Genetic Regulation Mechanisms for Type 2 Diabetes”, Xue et al 2018
- “Better Estimation of SNP Heritability from Summary Statistics Provides a New Understanding of the Genetic Architecture of Complex Traits”, Speed & Balding 2018
- “The Nature of Nurture: Using a Virtual-Parent Design to Test Parenting Effects on Children’s Educational Attainment in Genotyped Families”, Bates et al 2018
- “Quantitative Analysis of Population-Scale Family Trees With Millions of Relatives”, Kaplanis et al 2018
- “Meta-Analysis of Genome-Wide Association Studies for Cattle Stature Identifies Common Genes That Regulate Body Size in Mammals”, Bouwman et al 2018
- “Genome-Wide Study Identifies 611 Loci Associated With Risk Tolerance and Risky Behaviors”, Linnér et al 2018
- “The Genomic Commons”, Contreras & Knoppers 2018
- “Genomic Risk Prediction of Coronary Artery Disease in Nearly 500,000 Adults: Implications for Early Screening and Primary Prevention”, Inouye et al 2018
- “Genetic Architecture of Gene Expression Traits across Diverse Populations”, Mogil et al 2018
- “Fine-Mapping of an Expanded Set of Type 2 Diabetes Loci to Single-Variant Resolution Using High-Density Imputation and Islet-Specific Epigenome Maps”, Mahajan et al 2018
- “Mixed Model Association for Biobank-Scale Data Sets”, Loh et al 2018
- “Genome-Wide Association Study of 1 Million People Identifies 111 Loci for Atrial Fibrillation”, Nielsen et al 2018
- “Generalizing Genetic Risk Scores from Europeans to Hispanics/Latinos”, Grinde et al 2018
- “The Shared Genetic Basis of Human Fluid Intelligence and Brain Morphology”, Ge et al 2018
- “A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease”, Abul-Husn et al 2018
- “Muscle Health and Performance in Monozygotic Twins With 30 Years of Discordant Exercise Habits”, Bathgate 2018
- “Joint Contributions of Rare Copy Number Variants and Common SNPs to Risk for Schizophrenia”, Bergen et al 2018
- “No Association between Urbanisation, Neighbourhood Deprivation and IBD: a Population-Based Study of 4 Million Individuals”
- “Runs of Homozygosity: Windows into Population History and Trait Architecture”, Ceballos et al 2018
- “Genome-Wide Mapping of Global-To-Local Genetic Effects on Human Facial Shape”, Claes et al 2018
- “Genotype Imputation from Large Reference Panels”, Das et al 2018
- “The Arrival of Social Science Genomics”, Freese 2018
- “Maternal Prenatal Depressive Symptoms and Risk for Early-Life Psychopathology in Offspring: Genetic Analyses in the Norwegian Mother and Child Birth Cohort Study”, Hannigan et al 2018
- “A Decade in Psychiatric GWAS Research”, Horwitz et al 2018
- “Genome-Wide Association Meta-Analysis of Individuals of European Ancestry Identifies New Loci Explaining a Substantial Fraction of Hair Color Variation and Heritability”, Hysi et al 2018
- “Exploring the Relationship between Polygenic Risk for Cannabis Use, Peer Cannabis Use and the Longitudinal Course of Cannabis Involvement”, Johnson et al 2018c
- “Genome-Wide Polygenic Scores for Common Diseases Identify Individuals With Risk Equivalent to Monogenic Mutations”, Khera et al 2018
- “Genetics of Blood Lipids among ~300,000 Multi-Ethnic Participants of the Million Veteran Program”, Klarin et al 2018
- “The Impact of Variation in Twin Relatedness on Estimates of Heritability and Environmental Influences”, Liu 2018
- “‘The Fixity of Whiteness’: Genetic Admixture and the Legacy of the One-Drop Rule”, Liz 2018
- “Refining the Accuracy of Validated Target Identification through Coding Variant Fine-Mapping in Type 2 Diabetes”, Mahajan et al 2018
- “Predicting Polygenic Risk of Psychiatric Disorders”, Martin et al 2018b
- “Genetic Influences on Musical Specialization: a Twin Study on Choice of Instrument and Music Genre”
- “Inferring Transmission Histories of Rare Alleles in Population-Scale Genealogies”, Nelson et al 2018
- “Relationship between Deleterious Variation, Genomic Autozygosity, and Disease Risk: Insights from The 1000 Genomes Project”, Pemberton & Szpiech 2018
- “The Biological Contributions to Gender Identity and Gender Diversity: Bringing Data to the Table”, Polderman 2018
- “Genetic Influence on Social Outcomes during and After the Soviet Era in Estonia”, Rimfeld et al 2018
- “Pairs of Genetically Unrelated Look-Alikes”, Segal et al 2018
- “GWAS for BMI: a Treasure Trove of Fundamental Insights into the Genetic Basis of Obesity”, Speakman et al 2018
- “Genome-Wide Association Meta-Analysis Highlights Light-Induced Signaling As a Driver for Refractive Error”, Tedja et al 2018
- “Developmental Origins of Chronic Physical Aggression: A Bio-Psycho-Social Model for the Next Generation of Preventive Interventions”, Tremblay et al 2018
- “Convergence of Placenta Biology and Genetic Risk for Schizophrenia”, Ursini et al 2018
- “Genetic Variation in Health Insurance Coverage”, Wehby & Shane 2018
- “Fine-Mapping and Functional Studies Highlight Potential Causal Variants for Rheumatoid Arthritis and Type 1 Diabetes”, Westra et al 2018
- “A Differential Evolution Approach to Feature Selection in Genomic Prediction”, Whalen 2018
- “Twin Classroom Dilemma: To Study Together or Separately?”
- “Identifying Loci Affecting Trait Variability and Detecting Interactions in Genome-Wide Association Studies”, Young et al 2018
- “Unravelling Quasi-Causal Environmental Effects via Phenotypic and Genetically Informed Multi-Rater Models: The Case of Differential Parenting and Authoritarianism”, Zapko-Willmes et al 2018
- “Genome-Wide Analyses Using UK Biobank Data Provide Insights into the Genetic Architecture of Osteoarthritis”, Zengini et al 2018
- “Estimation of Complex Effect-Size Distributions Using Summary-Level Statistics from Genome-Wide Association Studies across 32 Complex Traits”, Zhang et al 2018c
- “The Genetics of Fruit Flavour Preferences”, Klee & Tieman 2018
- “The Personal and Clinical Utility of Polygenic Risk Scores”, Torkamani et al 2018
- “Ideology Between the Lines”, Hannikainen 2018
- “Social and Genetic Pathways in Multigenerational Transmission of Educational Attainment”, Liu 2018b
- “Personality and Genetic Associations With Military Service”, Miles & Haider-Markel 2018
- “Sizing up Whole-Genome Sequencing Studies of Common Diseases”, Wray 2018b
- “The New Genetics of Intelligence”, Plomin & Stumm 2018
- “86 Genomic Sites Associated With Educational Attainment Provide Insight into the Biology of Cognitive Performance”, Lee 2018
- “Misestimation of Heritability and Prediction Accuracy of Male-Pattern Baldness”, Yap 2018
- “Revisiting the Children-Of-Twins Design: Improving Existing Models for the Exploration of Intergenerational Associations”, McAdams et al 2018
- “Relatedness Disequilibrium Regression Estimates Heritability without Environmental Bias”, Young et al 2018
- “The Genetics of University Success”, Smith-Woolley et al 2018
- “DeepGS: Predicting Phenotypes from Genotypes Using Deep Learning”, Ma et al 2017
- “The Nature of Nurture: Effects of Parental Genotypes”, Kong et al 2017
- “Estimating Heritability without Environmental Bias”, Young et al 2017
- “Implicit Causal Models for Genome-Wide Association Studies”, Tran & Blei 2017
- “Socioeconomic Status and Genetic Influences on Cognitive Development”, Figlioa et al 2017
- “Genome-Wide Meta-Analysis Associates HLA-DQA1/DRB1 and LPA and Lifestyle Factors With Human Longevity”, Joshi et al 2017
- “Genetic Analysis of over One Million People Identifies 535 Novel Loci for Blood Pressure”, Evangelou et al 2017
- “Biological Insights Into Muscular Strength: Genetic Findings in the UK Biobank”, Tikkanen et al 2017
- “Accurate Genomic Prediction Of Human Height”, Lello et al 2017
- “Multiethnic Meta-Analysis Identifies New Loci for Pulmonary Function”, Wyss et al 2017
- “Genetic Diversity Turns a New PAGE in Our Understanding of Complex Traits”, Wojcik et al 2017
- “Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register”, Hilker et al 2017
- “Genome-Wide Association Study of Habitual Physical Activity in over 277,000 UK Biobank Participants Identifies Novel Variants and Genetic Correlations With Chronotype and Obesity-Related Traits”, Klimentidis et al 2017
- “The Genetic Basis of Human Brain Structure and Function: 1,262 Genome-Wide Associations Found from 3,144 GWAS of Multimodal Brain Imaging Phenotypes from 9,707 UK Biobank Participants”, Elliott et al 2017
- “Identifying Genetic Variants That Affect Viability in Large Cohorts”, Mostafavi et al 2017
- “Genome-Wide Genetic Data on ~500,000 UK Biobank Participants”, Bycroft et al 2017
- “Statistical Correction of the Winner’s Curse Explains Replication Variability in Quantitative Trait Genome-Wide Association Studies”, Palmer & Pe’er 2017
- “10 Years of GWAS Discovery: Biology, Function, and Translation”, Visscher et al 2017
- “Sexual Dimorphism in the Genetic Influence on Human Childlessness”, Verweij et al 2017
- “Targeting Aggression in Severe Mental Illness: The Predictive Role of Genetic, Epigenetic, and Metabolomic Markers”, Manchia & Fanos 2017
- “Discovery of the First Genome-Wide Statistically-Large Risk Loci for ADHD”, Demontis et al 2017
- “Codebreaker: A Deeply Personal Quest Made Matthew Might a Leader in Precision Medicine and Brought Him to UAB”
- “Genetic Predisposition to Obesity and Medicare Expenditures”, Wehby et al 2017
- “Father Absence and Accelerated Reproductive Development”, Gaydosh et al 2017
- “Comparison of Methods That Use Whole Genome Data to Estimate the Heritability and Genetic Architecture of Complex Traits”, Evans et al 2017
- “Psychiatric Genomics: An Update and an Agenda”, Sullivan et al 2017
- “Biogeographic Ancestry and Socioeconomic Outcomes in the Americas: A Meta-Analysis”, Kirkegaard et al 2017
- “Analysis of Genetic Similarity among Friends and Schoolmates in the National Longitudinal Study of Adolescent to Adult Health (Add Health)”, Domingue et al 2017
- “Personalized Media: A Genetically-Informative Investigation of Individual Differences in Online Media Use”, Ayorech et al 2017
- “Genetic and Environmental Influences on Household Financial Distress”, Xu et al 2017
- “Covariate Selection for Association Screening in Multiphenotype Genetic Studies”, Aschard et al 2017b
- “A Genetic Basis of Economic Egalitarianism”, Batricevic & Littvay 2017
- “Copy Number Variation in Syndromic Forms of Psychiatric Illness: The Emerging Value of Clinical Genetic Testing in Psychiatry”
- “Co-Aggregation of Major Psychiatric Disorders in Individuals With First-Degree Relatives With Schizophrenia: a Nationwide Population-Based Study”, Cheng et al 2017
- “Predictive Accuracy of Combined Genetic and Environmental Risk Scores”
- “Little Evidence That Socioeconomic Status Modifies Heritability of Literacy and Numeracy in Australia”
- “Internet Addiction and Its Facets: The Role of Genetics and the Relation to Self-Directedness”, Hahn et al 2017
- “National Clinical Audit Data Decodes the Genetic Architecture of Developmental Dysplasia of the Hip”, Hatzikotoulas et al 2017
- “Effect of Sequence Variants on Variance in Glucose Levels Predicts Type 2 Diabetes Risk and Accounts for Heritability”, Ivarsdottir et al 2017
- “Inference on the Genetic Basis of Eye and Skin Color in an Admixed Population via Bayesian Linear Mixed Models”
- “Polygenic Scores via Penalized Regression on Summary Statistics”
- “Differences in Genetic and Environmental Variation in Adult Body Mass Index by Sex, Age, Time Period, and Region: an Individual-Based Pooled Analysis of 40 Twin Cohorts”
- “Protein-Altering Variants Associated With Body Mass Index Implicate Pathways That Control Energy Intake and Expenditure in Obesity”, Turcot et al 2017
- “2017 Constance Holden Memorial Address: Liberal Creationism”
- “Polygenic Prediction of Obsessive Compulsive Symptoms”, Zilhão et al 2017
- “Racial Minority Group Interest in Direct-To-Consumer Genetic Testing: Findings from the PGen Study”, Landry et al 2017
- “Beyond Questionable Research Methods: The Role of Omitted Relevant Research in the Credibility of Research”, Schmidt 2017
- “Twin Spouses and Unrelated Look-Alikes: New Views”, Segal 2017
- “Heritability of Working in a Creative Profession”, Roeling et al 2017
- “The Heritability of Autism Spectrum Disorder”, Sandin et al 2017
- “Associations of Coffee Genetic Risk Scores With Coffee, Tea and Other Beverages in the UK Biobank”, Taylor & Munafò 2016
- “Genetic Variation in the Social Environment Contributes to Health and Disease”, Baud et al 2016
- “Statistical Properties of Simple Random-Effects Models for Genetic Heritability”, Steinsaltz et al 2016
- “Genome-Wide Meta-Analysis of Cognitive Empathy: Heritability, and Correlates With Sex, Neuropsychiatric Conditions and Brain Anatomy”, Warrier et al 2016
- “Evidence That Lower Socioeconomic Position Accentuates Genetic Susceptibility to Obesity”, Tyrrell et al 2016
- “Genome-Wide Association Study of Antisocial Personality Disorder”, Rautiainen et al 2016
- “Genetic Prediction of Male Pattern Baldness”, Hagenaars et al 2016
- “How Cognitive Genetic Factors Influence Fertility Outcomes: A Mediational SEM Analysis”, Menie et al 2016
- “Phenome-Wide Heritability Analysis of the UK Biobank”, Ge et al 2016
- “Estimate of Disease Heritability Using 4.7 Million Familial Relationships Inferred from Electronic Health Records”, Polubriaginof et al 2016
- “Genome-Wide Variants of Eurasian Facial Shape Differentiation and DNA Based Face Prediction”, Qiao et al 2016
- “Cost-Effectiveness of Pharmacogenetic-Guided Treatment: Are We There Yet?”, Verbelen et al 2016
- “Genome-Wide Association Study Reveals Multiple Loci Influencing Normal Human Facial Morphology”, Shaffer et al 2016
- “Genome-Wide Association Study Identifies 74 Loci Associated With Educational Attainment”, Okbay et al 2016
- “Mega-Analysis of 31,396 Individuals from 6 Countries Uncovers Strong Gene-Environment Interaction for Human Fertility”, Tropf et al 2016
- “The Great Migration and African-American Genomic Diversity”, Baharian et al 2016
- “Mortality Selection in a Genetic Sample and Implications for Association Studies”, Domingue et al 2016
- “Genome-Wide Estimates of Heritability for Social Demographic Outcomes”, Domingue et al 2016
- “Molecular Genetic Contributions to Social Deprivation and Household Income in UK Biobank (n = 112,151)”, Hill et al 2016
- “Older Fathers’ Children Have Lower Evolutionary Fitness across Four Centuries and in Four Populations”, Arslan et al 2016
- “How Does a Cowbird Learn To Be a Cowbird? New Research Explains How These Brood Parasites—Who Are Raised by Other Species—Still Manage to Become Cowbirds”, Soniak 2016
- “Estimating Effect Sizes and Expected Replication Probabilities from GWAS Summary Statistics”, Holland et al 2016
- “The Evolutionary Genetics of Personality Revisited”, Penke & Jokela 2016
- “Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data”, Shi et al 2016
- “Paraphilic Sexual Interests and Sexually Coercive Behavior: A Population-Based Twin Study”, Baur et al 2016
- “Genome-Wide Association Study of Working Memory Brain Activation”, Blokland et al 2016
- “Developing and Evaluating Polygenic Risk Prediction Models for Stratified Disease Prevention”, Chatterjee et al 2016
- “Familial Aggregation of Attention-Deficit/hyperactivity Disorder”, Chen et al 2016
- “Genetic and Environmental Parent–Child Transmission of Value Orientations: An Extended Twin Family Study”
- “The MC1R Gene and Youthful Looks”, Liu et al 2016
- “Heritability and Causal Reasoning”, Lynch 2016
- “A Genetic Epidemiological Mega Analysis of Smoking Initiation in Adolescents”, Murugadoss 2016
- “School Achievement and Risk of Eating Disorders in a Swedish National Cohort”, Sundquist et al 2016
- “GWAS for Executive Function and Processing Speed Suggests Involvement of the CADM2 Gene”, Ibrahim-Verbaas et al 2016
- “PHENIX: A Multiple-Phenotype Imputation Method for Genetic Studies”, Dahl et al 2016
- “A Novel Sibling-Based Design to Quantify Genetic and Shared Environmental Effects: Application to Drug Abuse, Alcohol Use Disorder and Criminal Behavior”, Kendler et al 2016
- “Chorionicity and Heritability Estimates from Twin Studies: The Prenatal Environment of Twins and Their Resemblance Across a Large Number of Traits”, Beijsterveldt et al 2016
- “Genetic and Environmental Influences on Food Preferences in Adolescence”, Smith et al 2016
- “Out on Their Own: a Test of Adult-Assisted Dispersal in Fledgling Brood Parasites Reveals Solitary Departures from Hosts”, Louder et al 2015
- “Heritability of Neuroanatomical Shape”, Ge et al 2015
- “An Empirical Bayes Mixture Model for Effect Size Distributions in Genome-Wide Association Studies”, Thompson et al 2015
- “Individual Esthetic Preferences for Faces Are Shaped Mostly by Environments, Not Genes”, Germine et al 2015
- “Meta-Analysis of Twin Studies Highlights the Importance of Genetic Variation in Primary School Educational Achievement”, Zeeuw et al 2015
- “SNP Hits on Cognitive Ability from 300k Individuals”, Hsu 2015
- “Genetic Variance Estimation With Imputed Variants Finds Negligible Missing Heritability for Human Height and Body Mass Index”, Yang et al 2015
- “Haplotypes of Common SNPs Can Explain Missing Heritability of Complex Diseases”, Bhatia et al 2015
- “The Support of Human Genetic Evidence for Approved Drug Indications”, Tipney et al 2015
- “Signaling and Productivity in the Private Financial Returns to Schooling”, Bingley et al 2015
- “Meta-Analysis of the Heritability of Human Traits Based on 50 Years of Twin Studies”, Polderman et al 2015
- “The Genetic Architecture of Pediatric Cognitive Abilities in the Philadelphia Neurodevelopmental Cohort”, Robinson et al 2015
- “Genetic and Environmental Influences on Obesity-Related Phenotypes in Chinese Twins Reared Apart and Together”, Zhou et al 2015
- “Is the Effect of Parental Education on Offspring Biased or Moderated by Genotype?”, Conley et al 2015
- “Genetic Contributions to Variation in General Cognitive Function: a Meta-Analysis of Genome-Wide Association Studies in the CHARGE Consortium (N = 53,949)”, Davies et al 2015
- “Is That Me or My Twin? Lack of Self-Face Recognition Advantage in Identical Twins”, Martini et al 2015
- “The Contribution of Additive Genetic Variation to Personality Variation: Heritability of Personality”, Dochtermann et al 2015
- “HERITABILITY STUDIES IN THE POSTGENOMIC ERA: THE FATAL FLAW IS CONCEPTUAL*”
- “Personality Related Traits As Predictors of Music Practice: Underlying Environmental and Genetic Influences”, Butkovic et al 2015
- “Heritability of Compulsive Internet Use in Adolescents”, Vink et al 2015
- “Physical Activity, Fitness, Glucose Homeostasis, and Brain Morphology in Twins”, Rottensteiner et al 2015
- “Genetic Studies of Body Mass Index Yield New Insights for Obesity Biology”, Locke et al 2015
- “Genome-Wide Meta-Analysis Identifies Six Novel Loci Associated With Habitual Coffee Consumption”, Cornelis et al 2015
- “Novel Loci Associated With Usual Sleep Duration: the CHARGE Consortium Genome-Wide Association Study”, Gottlieb et al 2015
- “Sexual Offending Runs in Families: A 37-Year Nationwide Study”, Långström et al 2015
- “LD Score Regression Distinguishes Confounding from Polygenicity in Genome-Wide Association Studies”, Bulik-Sullivan et al 2015
- “LDpred: Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores”, Vilhjálmsson et al 2015
- “The Fine-Scale Genetic Structure of the British Population”, Leslie et al 2015
- “Multi-Ancestry Genome-Wide Association Study of 21,000 Cases and 95,000 Controls Identifies New Risk Loci for Atopic Dermatitis”, Paternoster et al 2015
- “Genetic Background of Extreme Violent Behavior”, Tiihonen et al 2015
- “Association Analyses Identify 38 Susceptibility Loci for Inflammatory Bowel Disease and Highlight Shared Genetic Risk across Populations”, Liu et al 2015
- “Genome-Wide Identification of MicroRNAs Regulating Cholesterol and Triglyceride Homeostasis”, Wagschal 2015
- “Common Polygenic Variation Enhances Risk Prediction for Alzheimer’s Disease”, Escott-Price et al 2015
- “The Intergenerational Transmission of Anxiety: A Children-Of-Twins Study”, Eley et al 2015
- “Shared Genetic Influences Between Attention-Deficit/Hyperactivity Disorder (ADHD) Traits in Children and Clinical ADHD”, Stergiakouli 2015
- “The Etiologic Role of Genetic and Environmental Factors in Criminal Behavior As Determined from Full & Half-Sibling Pairs: an Evaluation of the Validity of the Twin Method”, Kendler et al 2014
- “Political Attitudes Develop Independently of Personality Traits”, Hatemi & Verhulst 2014
- “Simultaneous Discovery, Estimation and Prediction Analysis of Complex Traits Using a Bayesian Mixture Model”, Moser et al 2014
- “Attention-Deficit/Hyperactivity Disorder Polygenic Risk Scores Predict Attention Problems in a Population-Based Sample of Children”, Groen-Blokhuis et al 2014
- “On the Genetic Architecture of Intelligence and Other Quantitative Traits”, Hsu 2014
- “Biological Insights from 108 Schizophrenia-Associated Genetic Loci”, Consortium 2014
- “Nature, Nurture, and Expertise”, Plomin et al 2014
- “Genetic Variation Associated With Differential Educational Attainment in Adults Has Anticipated Associations With School Performance in Children”, Ward et al 2014
- “Heritability of Creative Achievement”, Piffer & Hur 2014
- “Results of a ‘GWAS Plus:’ General Cognitive Ability Is Substantially Heritable and Massively Polygenic”, Kirkpatrick et al 2014
- “Pulling Back The Curtain On Heritability Studies: Biosocial Criminology In The Postgenomic Era”, Burt & Simons 2014
- “Genome-Wide Association Study of Proneness to Anger”, Mick et al 2014
- “Genetic and Environmental Influences on Problematic Internet Use: A Twin Study”, Deryakulu & Ursavaş 2014
- “Social Influence Constrained by the Heritability of Attitudes”, Schwab 2014
- “Uncovering the Hidden Risk Architecture of the Schizophrenias: Confirmation in 3 Independent Genome-Wide Association Studies”, Arnedo 2014
- “The Genetics of Music Accomplishment: Evidence for Gene-Environment Correlation and Interaction”, Hambrick & Tucker-Drob 2014
- “Understanding the Relative Contributions of Direct Environmental Effects and Passive Genotype-Environment Correlations in the Association between Familial Risk Factors and Child Disruptive Behavior Disorders”, Bornovalova et al 2014
- “Genetics of Rheumatoid Arthritis Contributes to Biology and Drug Discovery”, Okada et al 2014
- “Genome-Wide Trans-Ancestry Meta-Analysis Provides Insight into the Genetic Architecture of Type 2 Diabetes Susceptibility”, Mahajan et al 2014
- “The Genetic Basis of Music Ability”, Tan et al 2014
- “Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies 6 New Risk Loci for Parkinson’s Disease”, Nalls et al 2014
- “Parent-Of-Origin-Specific Allelic Associations among 106 Genomic Loci for Age at Menarche”, Perry et al 2014
- “Defining the Role of Common Variation in the Genomic and Biological Architecture of Adult Human Height”, Wood et al 2014
- “A Genome-Wide Association Study of Anorexia Nervosa”, Boraska et al 2014
- “Replicability and Robustness of Genome-Wide-Association Studies for Behavioral Traits”, Rietveld et al 2014
- “Impact of Measurement Error on Testing Genetic Association With Quantitative Traits”, Liao et al 2013
- “Heritability and the Equal Environments Assumption: Evidence from Multiple Samples of Misclassified Twins”, Conley et al 2013
- “GWAS of 126,559 Individuals Identifies Genetic Variants Associated With Educational Attainment”, Rietveld et al 2013
- “Using Extended Genealogy to Estimate Components of Heritability for 23 Quantitative and Dichotomous Traits”, Zaitlen et al 2013
- “Childhood Intelligence Is Heritable, Highly Polygenic and Associated With FNBP1L”, Benyamin et al 2013
- “Genetic Architecture of Skin and Eye Color in an African-European Admixed Population”, Beleza et al 2013
- “An Opposite-Direction Modulation of the COMT Val158Met Polymorphism on the Clinical Response to Intrathecal Morphine and Triptans”, Cargnin et al 2013
- “Common DNA Variants Predict Tall Stature in Europeans”, Liu et al 2013
- “Contribution of Common Genetic Variants to Antidepressant Response”, Tansey et al 2013
- “Should Evolutionary Geneticists Worry about Higher-Order Epistasis?”, Weinreich et al 2013
- “Unrelated Look-Alikes: Replicated Study of Personality Similarity and Qualitative Findings on Social Relatedness”
- “Personality Similarity in Unrelated Look-Alike Pairs: Addressing a Twin Study Challenge”
- “The Wilson Effect: The Increase in Heritability of IQ With Age”
- “Http://www.science.sciencemag.org/highwire/filestream/594571/field_highwire_adjunct_files/1/Rietveld.SM.revision.2.pdf”
- “The Swedish Twin Registry: Establishment of a Biobank and Other Recent Developments”, Magnusson 2013
- “Genetic Risk Factors for BMI and Obesity in an Ethnically Diverse Population: Results from the Population Architecture Using Genomics and Epidemiology (PAGE) Study”, Fesinmeyer et al 2013
- “Tests of a Direct Effect of Childhood Abuse on Adult Borderline Personality Disorder Traits: a Longitudinal Discordant Twin Design”, Bornovalova et al 2013
- “Common DNA Markers Can Account for More Than Half of the Genetic Influence on Cognitive Abilities”, Plomin et al 2013
- “Large-Scale Association Analysis Identifies New Risk Loci for Coronary Artery Disease”, Deloukas et al 2013
- “A Meta-Analysis Identifies New Loci Associated With Body Mass Index in Individuals of African Ancestry”, Monda et al 2013
- “Critical Need for Family-Based, Quasi-Experimental Designs in Integrating Genetic and Social Science Research”, D’Onofrio et al 2013
- “A Genome-Wide Association Study of Behavioral Disinhibition”, McGue et al 2013
- “Genome-Wide Meta-Analysis Identifies 11 New Loci for Anthropometric Traits and Provides Insights into Genetic Architecture”, Berndt et al 2013
- “Large-Scale Genotyping Identifies a New Locus at 22q13.2 Associated With Female Breast Size”, Li et al 2013
- “The Half-Life of DNA in Bone: Measuring Decay Kinetics in 158 Dated Fossils”, Allentoft et al 2012
- “Genetics of Aggression”, Anholt & Mackay 2012
- “Unraveling the Genetic Etiology of Adult Antisocial Behavior: A Genome-Wide Association Study”, Tielbeek et al 2012
- “The Continuing Value of Twin Studies in the Omics Era”, Dongen et al 2012
- “Correlation and Causation in the Study of Personality”, Lee 2012
- “Genetic and Environmental Influences on Media Use and Communication Behaviors”, Kirzinger et al 2012
- “Sex Differences in Educational Attainment”, Mikk et al 2012
- “The Heritability and Genetic Correlates of Mobile Phone Use: a Twin Study of Consumer Behavior”, Miller et al 2012
- “Fullerton Virtual Twin Study: An Update”, Segal et al 2012
- “Relationship between Adiposity and Admixture in African-American and Hispanic-American Women”, Nassir et al 2012
- “Five Years of GWAS Discovery”, Visscher et al 2012
- “Large-Scale Association Analysis Provides Insights into the Genetic Architecture and Pathophysiology of Type 2 Diabetes”, Morris et al 2012
- “Effect of Shared Environmental Factors on Exercise Behavior from Age 7 to 12 Years”, Huppertz et al 2012
- “The Heritability of Avoidant and Dependent Personality Disorder Assessed by Personal Interview and Questionnaire”, Gjerde et al 2012
- “Most Reported Genetic Associations With General Intelligence Are Probably False Positives”, Chabris et al 2012
- “Genetic Variants and Associations of 25-Hydroxyvitamin D Concentrations With Major Clinical Outcomes”, Levin et al 2012
- “Research in China on the Molecular Genetics of Schizophrenia”, Cui & Jiang 2012
- “Genetic and Environmental Influences on Individual Differences in Frequency of Play With Pets among Middle-Aged Men: A Behavioral Genetic Analysis”, Jacobson et al 2012
- “Genomic Ancestry, Self-Reported ‘Color’ and Quantitative Measures of Skin Pigmentation in Brazilian Admixed Siblings”, Leite et al 2011
- “Reconsidering the Heritability of Intelligence in Adulthood: Taking Assortative Mating and Cultural Transmission into Account”, Vinkhuyzen et al 2011
- “A Novel, Functional and Replicable Risk Gene Region for Alcohol Dependence Identified by Genome-Wide Association Study”, Zuo et al 2011
- “Large-Scale Genome-Wide Association Analysis of Bipolar Disorder Identifies a New Susceptibility Locus near ODZ4”, Sklar et al 2011
- “Genome-Wide Association Studies Establish That Human Intelligence Is Highly Heritable and Polygenic”, Davies et al 2011
- “The Story of Emily and Control”, Alexander 2011
- “The Biological Roots of Complex Thinking: Are Heritable Attitudes More Complex?”, Conway et al 2011
- “Exercise Participation in Adolescents and Their Parents: Evidence for Genetic and Generation Specific Environmental Effects”, Moor et al 2011
- “The Etiology of Stability and Change in Religious Values and Religious Attendance”, Button et al 2011
- “Variation in Actual Relationship As a Consequence of Mendelian Sampling and Linkage”, Hill & Weir 2011
- “Genome-Wide Association Study of the Child Behavior Checklist Dysregulation Profile”, Mick et al 2011
- “Alcohol Dependence in Men: Reliability and Heritability”, Ystrom et al 2011
- “A Genome-Wide Association Study of Aging”, Walter et al 2011
- “A Critical Review of the First 10 Years of Candidate Gene-By-Environment Interaction Research in Psychiatry”, Duncan & Keller 2011
- “Meta-Analysis of Genome-Wide Association Studies of Asthma in Ethnically Diverse North American Populations”, Torgerson et al 2011
- “Genome-Wide Association Study of Conduct Disorder Symptomatology”, Dick et al 2011
- “Estimating the Total Number of Susceptibility Variants Underlying Complex Diseases from Genome-Wide Association Studies”, So et al 2010
- “On the Heritability of Consumer Decision Making: An Exploratory Approach for Studying Genetic Effects on Judgment and Choice”, Simonson & Sela 2010
- “Consistent Association of Type 2 Diabetes Risk Variants Found in Europeans in Diverse Racial and Ethnic Groups”, Waters et al 2010
- “In Search of Genes Associated With Risk for Psychopathic Tendencies in Children: a Two-Stage Genome-Wide Association Study of Pooled DNA”, Viding et al 2010
- “Not by Twins Alone: Using the Extended Family Design to Investigate Genetic Influence on Political Beliefs”, Hatemi et al 2010
- “Environmental Influences on Children’s Physical Activity: Quantitative Estimates Using a Twin Design”, Fisher et al 2010
- “A Longitudinal Study on Genetic and Environmental Influences on Leisure Time Physical Activity in the Finnish Twin Cohort”, Aaltonen et al 2010
- “No Effect of Classroom Sharing on Educational Achievement in Twins: a Prospective, Longitudinal Cohort Study”, Polderman et al 2010
- “Admixture Mapping Comes of Age”, Winkler et al 2010
- “Human Face Recognition Ability Is Specific and Highly Heritable”, Wilmer et al 2010
- “On Epistasis: Why It Is Unimportant in Polygenic Directional Selection”, Crow 2010
- “Genetic and Environmental Influences on the Transmission of Parental Depression to Children’s Depression and Conduct Disturbance: an Extended Children of Twins Study”, Silberg et al 2010
- “Effects of Modafinil on the Sleep EEG Depend on Val158Met Genotype of COMT”, Bodenmann & Landolt 2010
- “Meta-Analysis of Genome-Wide Association Studies of Attention-Deficit/hyperactivity Disorder”, Neale et al 2010
- “Blood Vitamin D Levels in Relation to Genetic Estimation of African Ancestry”, Signorello et al 2010
- “Hundreds of Variants Clustered in Genomic Loci and Biological Pathways Affect Human Height”, Allen et al 2010
- “Common SNPs Explain a Large Proportion of the Heritability for Human Height”, Yang et al 2010
- “Genetic Ancestry, Social Classification, and Racial Inequalities in Blood Pressure in Southeastern Puerto Rico”, Gravlee et al 2009
- “Genetics of Human Aggressive Behavior”, Craig & Halton 2009
- “The Heritability of General Cognitive Ability Increases Linearly from Childhood to Young Adulthood”, Haworth et al 2009
- “Musical Aptitude Is Associated With AVPR1A-Haplotypes”, Ukkola et al 2009
- “Factors Contributing to the Facial Aging of Identical Twins”, Guyuron et al 2009
- “Material Values Are Largely in the Family: A Twin Study of Genetic and Environmental Contributions to Materialism”, Giddens et al 2009
- “History of Behavior Genetics”, Loehlin 2009
- “Genetics of Exercise Behavior”, Stubbe & Geus 2009
- “Nature versus Nurture Is Nonsense: On the Necessity of an Integrated Genetic, Social, Developmental, and Personality Psychology”, Barlow 2009
- “Association of Substance Use Disorders With Childhood Trauma but Not African Genetic Heritage in an African American Cohort”, Ducci et al 2009
- “Parental Alcoholism and Offspring Behavior Problems: Findings in Australian Children of Twins”, Waldron et al 2009
- “Sexy Sons and Sexy Daughters: the Influence of Parents’ Facial Characteristics on Offspring”, Cornwell & Perrett 2008
- “Unique Environmental Effects on Physical Activity Participation: A Twin Study”, Duncan et al 2008
- “Transmission of Attitudes Toward Abortion and Gay Rights: Effects of Genes, Social Learning and Mate Selection”, Eaves & Hatemi 2008b
- “Heritability in the Genomics Era—Concepts and Misconceptions”, Visscher et al 2008
- “Data and Theory Point to Mainly Additive Genetic Variance for Complex Traits”, Hill et al 2008
- “A Panel of Ancestry Informative Markers for Estimating Individual Biogeographical Ancestry and Admixture from 4 Continents: Utility and Applications”
- “A Twin-Family Study of General IQ”, Leeuwen et al 2008
- “Genetic Mapping in Human Disease”, Altshuler et al 2008
- “Origins of Magic: Review of Genetic and Epigenetic Effects”, Ramagopalan et al 2007
- “The Genetics of Political Attitudes”, Hatemi 2007
- “A Children of Twins Study of Parental Divorce and Offspring Psychopathology”, D’Onofrio et al 2007
- “A Common Variant of HMGA2 Is Associated With Adult and Childhood Height in the General Population”, Weedon et al 2007
- “Genome Partitioning of Genetic Variation for Height from 11,214 Sibling Pairs”, Visscher 2007
- “The Evolution of Personality Variation in Humans and Other Animals”, Nettle 2006
- “Achievement and Ascription in Educational Attainment: Genetic and Environmental Influences on Adolescent Schooling”, Nielsen 2006
- “Genetic and Cultural Transmission of Smoking Initiation: An Extended Twin Kinship Model”, Maes et al 2006
- “Two Monozygotic Twin Pairs Discordant for Female-To-Male Transsexualism”, Segal 2006
- “Smarter Than The Average Bug”, McCrone 2006
- “Psychological Essentialism and Stereotype Endorsement”, Bastian & Haslam 2006
- “Assumption-Free Estimation of Heritability from Genome-Wide Identity-By-Descent Sharing between Full Siblings”, Visscher et al 2006
- “Influence of Environmental Factors on Facial Aging”, Rexbye et al 2006
- “Genetic Determinism”, Anonymous 2006
- “Harry Potter and the Recessive Allele”, Craig et al 2005
- “Nature and Nurture: Genetic and Environmental Influences on Behavior”, Plomin & Asbury 2005b
- “Sports Participation during Adolescence: A Shift from Environmental to Genetic Factors”, Stubbe et al 2005
- “Personality Traits As Intermediary Phenotypes in Suicidal Behavior: Genetic Issues”, Baud 2005
- “Contributions of Parental Alcoholism, Prenatal Substance Exposure, and Genetic Transmission to Child ADHD Risk: A Female Twin Study”, Knopik et al 2005
- “Genetic and Environmental Influences on Antisocial Behaviors: Evidence from Behavioral-Genetic Research”, Moffitt 2005
- “Under the Skin: On the Impartial Treatment of Genetic and Environmental Hypotheses of Racial Differences”, Rowe 2005
- “Personality Development: Stability and Change”, Caspi 2005
- “The Cognitive Cost of Being a Twin: Evidence from Comparisons within Families in the Aberdeen Children of the 1950s Cohort Study”, Ronalds 2005
- “Neurobiology of Intelligence: Science and Ethics”, Gray & Thompson 2004
- “Intelligence: Genetics, Genes, and Genomics”, Plomin & Spinath 2004
- “Genetic and Environmental Effects on Offspring Alcoholism: New Insights Using an Offspring-Of-Twins Design”, Jacob et al 2003
- “Genetics and Educational Psychology”
- “A Model-Fitting Implementation of the DeFries-Fulker Model for Selected Twin Data”
- “The Influence of Genetic Factors on Physical Functioning and Exercise in Second Half of Life”
- “Genetic Factors in Physical Activity Levels: A Twin Study”, Maia et al 2002
- “Selection, Structure and the Heritability of Behavior”, Stirling et al 2002
- “A Genome-Wide Scan of 1842 DNA Markers for Allelic Associations With General Cognitive Ability: A Five-Stage Design Using DNA Pooling and Extreme Selected Groups”, Plomin et al 2001
- “Individual Differences in Response to Regular Physical Activity”, Bouchard & Rankinen 2001
- “Heritability Estimates versus Large Environmental Effects: The IQ Paradox Resolved”, Dickens & Flynn 2001
- “Nonshared Environment: A Theoretical, Methodological, and Quantitative Review”, Turkheimer & Waldron 2000
- “Three Laws of Behavior Genetics and What They Mean”, Turkheimer 2000
- “Early Observations of Genetic Diseases”, Urban 1999
- “Comparing the Biological and Cultural Inheritance of Personality and Social Att”, Eaves et al 1999
- “Causality in Complex Systems”, Wagner 1999
- “Chapter 25: Liability-Threshold Model”, Lynch & Walsh 1998
- “Poverty and Behavior: Are Environmental Measures Nature and Nurture?”, Rowe & Rodgers 1997
- “Francis Galton: His Approach to Polygenic Disease”, Galton & Galton 1997
- “An Examination of Genotype-Environment Interactions for Academic Achievement in an US National Longitudinal Survey”, Ord & Rowe 1997
- “A ‘Second Front’ in Soviet Genetics: The International Dimension of the Lysenko Controversy, 1944–1947”, Krementsov 1996
- “Population Stratifications Can Cause False Positive Linkage Results If Founders Are Untyped”
- “Barbara Stoddard Burks: Pioneer Behavioral Geneticist and Humanitarian [Ch15]”, King et al 1996
- “Galton Lecture: Behavior Genetic Studies of Intelligence, Yesterday and Today: the Long Journey from Plausibility to Proof”, Bouchard 1996
- “Arguments About Aborigines: Australia and the Evolution of Social Anthropology: Chapter 7, Conceptions and Misconceptions”, Hiatt 1996
- “Conceptions and Misconceptions § Difficulties in Understanding Sex”, Hiatt 1996 (page 21)
- “Separate Social Worlds of Siblings”, Hetherington et al 1994
- “Smoking and Sports Participation”, Koopmans et al 1994
- “Nature-Nurture Reconceptualized in Developmental Perspective: A Bioecological Model”, Bronfenbrenner & Ceci 1994
- “The Lifetime History of Major Depression in Women: Reliability of Diagnosis and Heritability”, Kendler et al 1993
- “Evolution of the Nature~nurture Issue in the History of Psychology”, PsycBooks 1993
- “Heritability of Interests: a Twin Study”, Lykken et al 1993
- “Dyslexia Subtypes, Genetics, Behavior, and Brain Imaging”, Lubs et al 1991
- “The Response to Long-Term Overfeeding in Identical Twins”, Bouchard et al 1990b
- “The Importance of Twin Studies for Individual Differences Research”
- “Confirming Unexpressed Genotypes for Schizophrenia: Risks in the Offspring of Fischer’s Danish Identical and Fraternal Discordant Twins”, Gottesman & Bertelsen 1989
- “Genetic and Environmental Determinants of Musical Ability in Twins”
- “Genes, Culture and Personality: An Empirical Approach”, Eaves et al 1989
- “Behavior Genetics Association Abstract”
- “Resemblances of Parents and Twins in Sports Participation and Heart Rate”, Boomsma et al 1989
- “Behavior Genetics Association Abstracts”
- “Offspring of Twin Pairs Discordant for Psychiatric Illness”, Bertelsen & Gottesman 1986
- “A Twin Study of Human Obesity”, Stunkard et al 1986b
- “Storm Over Biology: Essays on Science, Sentiment, and Public Policy”, Davis et al 1986
- “Multiple Regression Analysis of Twin Data Obtained from Selected Samples”
- “The Study of Temperament: Changes, Continuities and Challenges”, Plomin & Dunn 1986
- “Geneticists and Race”, Provine 1986
- “Temperament (PLE: Emotion): Early Developing Personality Traits”, Buss et al 1984
- “The Colorado Adoption Project”
- “Identical Twins Reared Apart: Reanalysis or Pseudo-Analysis?”, Bouchard 1982
- “Genetic and Environmental Influences on the Development of Piagetian Logico-Mathematical Concepts and Other Specific Cognitive Abilities: A Twin Study”, Garfinkle 1982
- “Cigarette Smoking, Use of Alcohol, and Leisure-Time Physical Activity among Same-Sexed Adult Male Twins”, Kaprio et al 1981
- “Twins: Black and White”, Osborne 1980
- “Personality and Heredity: An Introduction to Psychogenetics”, Wells 1980
- “A Note on the Heritability of Memory Span”
- “Twin Method: Defense of a Critical Assumption”, Scarr & Carter-Saltzman 1979
- “Infantile Autism: A Genetic Study Of 21 Twin Pairs”, Folstein & Rutter 1977
- “Social Genetics”
- “Heritability of IQ [Letter in Response to ‘The Heritability Hang-Up’, by M. Feldman and R. C. Lewontin]”, Plomin & DeFries 1976
- “The Multifactorial/threshold Concept-Uses and Misuses”
- “Issues in the Genetics of Social Behavior”
- “Sibling Deidentification”, Schachter et al 1976
- “The Determinants of Earnings: Genetics, Family, and Other Environments: A Study of White Male Twins”
- “The Professor, The Institute and DNA”, Dubos 1976
- Heredity, Environment, & Personality: A Study of 850 Sets of Twins, Loehlin & Nichols 1976
- “The Meaning of Heritability in the Behavioral Sciences”, Jensen 1975b
- “A Test of a Primary Bias in Twin Studies with respect to Measured Ability”
- “Biological Aspects of a High Socio-Economic Group II. IQ Components and Social Mobility”, Gibson & Mascie-Taylor 1973b
- “The Multifactorial Model for the Inheritance of Liability to Disease and Its Implications for Relatives at Risk”
- Genetics, Environment, and Behavior: Implications for Educational Policy, Ehrman et al 1972
- “Behavior and Heredity [Open Letter]”, Page 1972c
- “Heritability of Liability and Concordance in Monozygous Twins”, Smith 1970
- “Schizophrenia: Evidence for the Major Gene Hypothesis”, Elston & Campbell 1970
- “Behavioral Genetics [Annual Review]”, McClearn 1970
- “Biology and Behavior: Genetics”, Glass 1968
- “The Effect of Assortative Mating on the Genetic Composition of a Population”, Crow & Felsenstein 1968
- “The Inheritance of Liability to Diseases With Variable Age of Onset, With Particular Reference to Diabetes Mellitus”, Falconer 1967
- “Hereditary Influences on Vocational Preferences As Shown by Scores of Twins on the Minnesota Vocational Interest Inventory”, Vandenberg & Stafford 1967
- “Possible Transfer of Metallurgical and Astronomical Approaches to Problem of Environment versus Ethnic Heredity”, Shockley 1966
- “Commentary on R. A. Fisher‘s Paper on 'The Correlation Between Relatives on the Supposition of Mendelian Inheritance’”, Moran & Smith 1966
- “Genetic Factors in Activity Motivation”
- “The Inheritance of Liability to Certain Diseases, Estimated from the Incidence among Relatives”
- “Hereditary Components in Vocational Preferences”, Vandenberg & Kelly 1964
- “Roots of Behavior: Genetics, Insect, and Socialization in Animal Behavior”, Bliss 1962
- “Behavior Genetics”, Fuller 1960
- “Heredity, Environment, and the Question "How?"”, Anastasi 1958
- “The Potential Contribution of Longitudinal Twin Studies: An Appraisal”, Falkner 1957
- “The Importance of Being Cross-Bred”, Michie & McLaren 1955
- Twins: A Study of 3 Pairs of Identical Twins, Burlingham 1952
- “Studies of Identical Twins Reared Apart”, Burks & Roe 1949
- “The Nature-Nurture Controversy”, Pastore 1949
- “The Incidence and Mendelian Transmission of Mid-Digital Hair in Man”, Bernstein & Burks 1942
- “Twins: A Study of Heredity and Environment”, Newman et al 1937
- “Twin-Similarities in Personality Traits”, Carter 1933
- “Statistical Methods in Biology”, Wright 1931
- “The Biological Basis of Human Nature”, Jennings 1930
- “Chapter 10: The Relative Influence of Nature and Nurture upon Mental Development: A Comparative Study of Foster Parent-Offspring Child Resemblance and True Parent-True Child Resemblance”, Burks 1928b
- “Chapter II: Statistical Hazards in Nature-Nurture Investigations”, Burks 1928
- “Heredity and Environment”, Jennings 1924
- “On the Inheritance of the Mental and Moral Characters in Man, and Its Comparison With the Inheritance of the Physical Characters”, Pearson 1903
- “A Mega-Analysis of Genome-Wide Association Studies for Major Depressive Disorder”
- “I’m Not A Look-A-Like!”, Brunelle 2026
- “The Lettre Lab: Members”, Lettre 2026
- “Behavior Genetic Frameworks of Causal Reasoning for Personality Psychology”
- “Applying Compressed Sensing to Genome-Wide Association Studies”
- “Determination of Nonlinear Genetic Architecture Using Compressed Sensing”
- “Common Genetic Variants Associated With Cognitive Performance”, Hsu 2026
- “Big Data: Astronomical or Genomical?”, Stephens et al 2026
- “G = E: What GWAS Can Tell Us about the Environment”, Gage et al 2026
- “Andrew Wood”
- “Genetic Determinants of the Gut Microbiome in UK Twins”
- “Gut Microbiome Heritability Is Near-Universal but Environmentally Contingent”
- “Jean-Baptiste Pingault”
- “The Right Not to Know: When Ignorance Is Bliss but Deadly”
- “Serena Sanna”
- “Society Is Fixed, Biology Is Mutable”
- “Is Bronny James Underrated? Inside the Phenomenon of the NBA Bloodline”
- “The Myth of Human Fragility”
- “Personality Polygenes, Positive Affect, and Life Satisfaction”
- “A Newly Discovered Tea Plant Is Caffeine-Free: It Was Found Growing Wild in Fujian Province”
- “DNA Sequencing Costs: Data”
- “The Cost of Sequencing a Human Genome”
- “Association of the Anxiogenic and Alerting Effects of Caffeine With ADORA2A and ADORA1 Polymorphisms and Habitual Level of Caffeine Consumption”
- “The Texas Twin Project”
- “Genome-Wide Association Studies of a Broad Spectrum of Antisocial Behavior”
- “Estimating the Heritability of Cognitive Traits across Dog Breeds Reveals Highly Heritable Inhibitory Control and Communication Factors”
- “A New Test Can Predict IVF Embryos’ Risk of Having a Low IQ: A New Genetic Test That Enables People Having IVF to Screen out Embryos Likely to Have a Low IQ or High Disease Risk Could Soon Become Available in the US”
- “$400 Million Investment Programme Positions Ireland for Global Leadership in Genomic Research and Advanced Life Sciences”
- “Additive Genetic Variation in Schizophrenia Risk Is Shared by Populations of African and European Descent”
- “The Infinitesimal Model: Definition, Derivation, and Implications”
- “Xist Ribonucleoproteins Promote Female Sex-Biased Autoimmunity”
- “Intelligence Indexes Generalist Genes for Cognitive Abilities”
- “Is There a Genetic Link to Being an Extremely Good Boy?”
- “Now You Can Sequence Your Whole Genome for Just $200”
- “The Importance of Heritability in Psychological Research: The Case of Attitudes”
- doctorveera
- Sort By Magic
collectibleseducation-metricssoviet-genetics genetic-disease historical-genetics lysenko-controversy medical-genetics genetics-historyunderstanding-biasdna-decaydisease-susceptibilitypopulation-geneticsfacial-agingdna-profilingpolygenic-riskrecessive-allelegenetic-admixturephenotypic-plasticityeducation-outcomes
- Wikipedia (8)
- Miscellaneous
- Bibliography
See Also
Gwern
“Does Mouse Utopia Exist?”, Gwern 2019
“The Power of Twins: The Scottish Milk Experiment”, Gwern 2016
“𝐺𝑒𝑛𝑖𝑢𝑠 𝑅𝑒𝑣𝑖𝑠𝑖𝑡𝑒𝑑 Revisited”, Gwern 2016
Links
“Population-Specific Polygenic Risk Scores for People of Han Chinese Ancestry”, Chen et al 2025
Population-specific polygenic risk scores for people of Han Chinese ancestry
“Within-Family Heritability Estimates for Behavioral and Disease Phenotypes from 500,000 Sibling Pairs of Diverse Ancestries”, Yengo et al 2025
“Direct Effect of Genetic Ancestry on Complex Traits in a Mexican Population”, Wang et al 2025
Direct effect of genetic ancestry on complex traits in a Mexican population
“Importance of Clinical, Laboratory, and Genetic Risk Factors for Incident CAD”, Bhattacharya et al 2025
Importance of Clinical, Laboratory, and Genetic Risk Factors for Incident CAD
“Polygenic Score Prediction Within and Between Sibling Pairs for Intelligence, Cognitive Abilities, and Educational Traits From Childhood to Early Adulthood”, Lin et al 2025
“Three Promising Directions in the Study of Intelligence With Genetic Methods”, Lee & Morris 2025b
Three Promising Directions in the Study of Intelligence With Genetic Methods
“Nature, Nurture, and Socioeconomic Outcomes: New Evidence from Sib Pairs and Molecular Genetic Data”, Markel et al 2025
Nature, nurture, and socioeconomic outcomes: New evidence from sib pairs and molecular genetic data
“A Litmus Test for Confounding in Polygenic Scores”, Smith et al 2025
“Trans-Ancestry Genome-Wide Study of Depression Identifies 697 Associations Implicating Cell Types and Pharmacotherapies”, Consortium 2025
“Within-Family GWAS Does Not Ameliorate the Decline in Prediction Accuracy across Populations”, Zhang & Conley 2024
Within-Family GWAS does not Ameliorate the Decline in Prediction Accuracy across Populations
“Genetically-Diverse Crowds Are Wiser”, Barneron et al 2024
“DNA and IQ: Big Deal or Much Ado about Nothing? A Meta-Analysis”, Oxley et al 2024
DNA and IQ: Big deal or much ado about nothing? A meta-analysis
“Dietary Restriction Impacts Health and Lifespan of Genetically Diverse Mice”, Francesco et al 2024
Dietary restriction impacts health and lifespan of genetically diverse mice
“Analysis of 3.6 Million Individuals Yields Minimal Evidence of Pairwise Genetic Interactions for Height”, Jabalameli et al 2024
“Effect of Disclosing a Polygenic Risk Score for Coronary Heart Disease on Adverse Cardiovascular Events: 10-Year Follow-Up of the MI-GENES Randomized Clinical Trial”, Naderian et al 2024
“Efficient BlockLASSO for Polygenic Scores With Applications to All of Us and UK Biobank”, Raben et al 2024
Efficient blockLASSO for Polygenic Scores with Applications to All of Us and UK Biobank
“A 30-Nation Investigation of Lay Heritability Beliefs”, Ferris et al 2024
“Star Trek Fan Leaves Behind a Collection Like No One Has Done Before: When Troy Nelson Died, His Shelves Were Filled to the Rafters With Memorabilia from the Popular Franchise. Soon, the Massive Collection Will Be Boldly Going, Going, Gone”, Deb 2024
“Predicting the Direction of Phenotypic Difference”, Gokhman et al 2024
“The Nurture of Nature and the Nature of Nurture: How Genes and Investments Interact in the Formation of Skills”, Houmark et al 2024
“Gene-Environment Correlation: The Role of Family Environment in Academic Development”, Zhou et al 2024
Gene-environment correlation: The role of family environment in academic development
“Refining the Impact of Genetic Evidence on Clinical Success”, Minikel et al 2023
“Genetic Influences on Educational Attainment Through the Lens of the Evolving Swedish Welfare State: A Cross-Level Gene-Environment Interaction Study Based on Polygenic Indices and Longitudinal Register Data”, Pettersson 2023
“Assessing the Impact of 20th Century Internal Migrations on the Genetic Structure of Estonia”, Kuznetsov et al 2023
Assessing the impact of 20th century internal migrations on the genetic structure of Estonia
“The Heritability of Ability Tilts”, Coyle et al 2023
“Multi-Ancestry Polygenic Mechanisms of Type 2 Diabetes Elucidate Disease Processes and Clinical Heterogeneity”, Smith et al 2023
“It Matters What and Where We Measure: Education and Ideology in a Swedish Twin Design”, Ahlskog 2023
It Matters What and Where We Measure: Education and Ideology in a Swedish Twin Design
“A Biobank-Scale Test of Marginal Epistasis Reveals Genome-Wide Signals of Polygenic Epistasis”, Fu et al 2023
A biobank-scale test of marginal epistasis reveals genome-wide signals of polygenic epistasis
“GWAS of Random Glucose in 476,326 Individuals Provide Insights into Diabetes Pathophysiology, Complications and Treatment Stratification”, Lagou et al 2023
“Population-Level Genetic Variation Shapes Generative Brain Mechanisms”, Monaghan et al 2023
Population-Level Genetic Variation Shapes Generative Brain Mechanisms
“Principles and Methods for Transferring Polygenic Risk Scores across Global Populations”, Kachuri et al 2023
Principles and methods for transferring polygenic risk scores across global populations
“Deep Learning Based Phenotyping of Medical Images Improves Power for Gene Discovery of Complex Disease”, Flynn et al 2023
“Nature and Nurture in Fussy Eating from Toddlerhood to Early Adolescence: Findings from the Gemini Twin Cohort”, Nas et al 2023
“Overcoming Attenuation Bias in Regressions Using Polygenic Indices”, Kippersluis et al 2023
Overcoming attenuation bias in regressions using polygenic indices
“Estimation of Indirect Genetic Effects and Heritability under Assortative Mating”, Young 2023
Estimation of indirect genetic effects and heritability under assortative mating
“Multi-Ancestry Genome-Wide Meta-Analysis of 56,241 Individuals Identifies LRRC4C, LHX5-AS1 and Nominates Ancestry-Specific Loci PTPRK, GRB14, and KIAA0825 As Novel Risk Loci for Alzheimer Disease: the Alzheimer Disease Genetics Consortium”, Rajabli et al 2023
“Rapid and Accurate Multi-Phenotype Imputation for Millions of Individuals”, Gu et al 2023
Rapid and accurate multi-phenotype imputation for millions of individuals
“The Relationship between Genotype- and Phenotype-Based Estimates of Genetic Liability to Human Psychiatric Disorders, in Practice and in Theory.”, Krebs et al 2023
“Do Polygenic Indices Capture ‘Direct’ Effects on Child Externalizing Behavior? Within-Family Analyses in Two Longitudinal Birth Cohorts”, Tanksley et al 2023
“Shaping Faces: Genetic and Epigenetic Control of Craniofacial Morphogenesis”, Selleri & Rijli 2023
Shaping faces: genetic and epigenetic control of craniofacial morphogenesis
“Gene-Environment Interplay in Early Life Cognitive Development”, Stumm et al 2023
Gene-environment interplay in early life cognitive development
“The Big (Genetic) Sort? Reassessing Migration Patterns and Their Genetic Imprint in the UK”, Furuya et al 2023
The Big (Genetic) Sort? Reassessing Migration Patterns and Their Genetic Imprint in the UK
“Identification and Analysis of Individuals Who Deviate from Their Genetically-Predicted Phenotype”, Hawkes et al 2023
Identification and analysis of individuals who deviate from their genetically-predicted phenotype
“A 12-Gene Pharmacogenetic Panel to Prevent Adverse Drug Reactions: an Open-Label, Multicenter, Controlled, Cluster-Randomized Crossover Implementation Study (PREPARE)”, Swen et al 2023
“Sharing GWAS Summary Statistics Results in More Citations”, Reales & Wallace 2023
“Phenotype Integration Improves Power and Preserves Specificity in Biobank-Based Genetic Studies of MDD”, Dahl et al 2023
“Genetic and Environmental Influences on Playing Video Games”, Hassan 2023
“15 Years of GWAS Discovery: Realizing the Promise”, Abdellaoui et al 2023
“Childhood Maltreatment and Mental Health Problems: A Systematic Review and Meta-Analysis of Quasi-Experimental Studies”, Baldwin et al 2023
“The Heritability of Pescetarianism and Vegetarianism”, Wesseldijk et al 2023
“PATH: Defining Ancestry, Heritability and Plasticity of Cellular Phenotypes in Somatic Evolution”, Schiffman et al 2022
PATH: Defining ancestry, heritability and plasticity of cellular phenotypes in somatic evolution
“PIGEON: Reimagining Gene-Environment Interaction Analysis for Human Complex Traits”, Miao et al 2022
PIGEON: Reimagining Gene-Environment Interaction Analysis for Human Complex Traits
“A Genetically Informed Registered Report on Adverse Childhood Experiences and Mental Health”, Baldwin et al 2022
A genetically informed Registered Report on adverse childhood experiences and mental health
“Polygenic Risk Scores for the Prediction of Cardiometabolic Disease”, O’Sullivan et al 2022
Polygenic risk scores for the prediction of cardiometabolic disease
“Genetic Diversity Fuels Gene Discovery for Tobacco and Alcohol Use”, Saunders et al 2022
Genetic diversity fuels gene discovery for tobacco and alcohol use
“Genetic Influences on Educational Achievement in Cross-National Perspective”, Baier et al 2022
Genetic Influences on Educational Achievement in Cross-National Perspective
“The APOE Locus Is Linked to Decline in General Cognitive Function: 20-Years Follow-Up in the Doetinchem Cohort Study”, Rietman et al 2022
“Polygenic Prediction of Molecular Traits Using Large-Scale Meta-Analysis Summary Statistics”, Pain et al 2022
Polygenic Prediction of Molecular Traits using Large-Scale Meta-analysis Summary Statistics
“Parental Transmission and the Importance of the (Noncausal) Effects of Education on Political Engagement: Missing the Forest for the Trees”, Rasmussen et al 2022
“Familial Concentration of Crime in a Digital Era: Criminal Behavior among Family Members of Cyber Offenders”, Weijer & Moneva 2022
“Two Pup Vocalization Types Are Genetically and Functionally Separable in Deer Mice”, Jourjine et al 2022
Two pup vocalization types are genetically and functionally separable in deer mice
“Correcting for Volunteer Bias in GWAS Uncovers Novel Genetic Variants and Increases Heritability Estimates”, Alten et al 2022
“Advances and Applications of Polygenic Scores for Coronary Artery Disease”, Patel & Khera 2022
Advances and Applications of Polygenic Scores for Coronary Artery Disease
“Heritable and Sex-Specific Variation in the Development of Social Behavior in a Wild Primate”, Lange et al 2022
Heritable and sex-specific variation in the development of social behavior in a wild primate
“Benefits and Barriers to Implementing Precision Preventive Care: Results of a National Physician Survey”, Vassy et al 2022
“Polygenic Scoring Accuracy Varies across the Genetic Ancestry Continuum in All Human Populations”, Ding et al 2022
Polygenic scoring accuracy varies across the genetic ancestry continuum in all human populations
“Sibling Variation in Phenotype and Genotype: Polygenic Trait Distributions and DNA Recombination Mapping With UK Biobank and IVF Family Data”, Lello et al 2022
“Improving on Polygenic Scores across Complex Traits Using Select and Shrink With Summary Statistics”, Tyrer et al 2022
Improving on polygenic scores across complex traits using select and shrink with summary statistics
“Extremely Sparse Models of Linkage Disequilibrium in Ancestrally Diverse Association Studies”, Nowbandegani et al 2022
Extremely sparse models of linkage disequilibrium in ancestrally diverse association studies
“What Does Heritability of Alzheimer’s Disease Represent?”, Baker et al 2022
“The Phenotype-Genotype Reference Map: Improving Biobank Data Science through Replication.”, Bastarache et al 2022
The Phenotype-Genotype Reference Map: Improving biobank data science through replication.
“Global Biobank Analyses Provide Lessons for Developing Polygenic Risk Scores across Diverse Cohorts”, Wang et al 2022
Global biobank analyses provide lessons for developing polygenic risk scores across diverse cohorts
“Asymmetrical Genetic Attributions for the Presence and Absence of Health Problems”, Lebowitz et al 2022
Asymmetrical genetic attributions for the presence and absence of health problems
“Concordance for Gender Dysphoria in Genetic Female Monozygotic (Identical) Triplets”, Kauffman et al 2022
Concordance for Gender Dysphoria in Genetic Female Monozygotic (Identical) Triplets
“Molecular Genetics and Mid-Career Economic Mobility”, Minard 2022
“With LeBron James Junior Nearly Grown, Could Dad’s Preposterous NBA Dream Become Reality? LeBron James’s Last NBA Goal Might Be to Play Long Enough to Take the Court With His Oldest Son. But Is Bronny James a Legit NBA Prospect, and What Would It Take for the Lakers to Turn Father and Son into Teammates?”, Sherman 2022
“Cost-Effectiveness of Polygenic Risk Scores to Guide Statin Therapy for Cardiovascular Disease Prevention”, Kiflen et al 2022
“A Polygenic Risk Score to Predict Sudden Cardiac Arrest in Patients With Coronary Artery Disease”, Porcu et al 2022
A polygenic risk score to predict sudden cardiac arrest in patients with coronary artery disease
“Health Care Utilization of Fine-Scale Identity by Descent Clusters in a Los Angeles Biobank”, Caggiano et al 2022
Health care utilization of fine-scale identity by descent clusters in a Los Angeles biobank
“From Mendel to Quantitative Genetics in the Genome Era: the Scientific Legacy of W. G. Hill”, Charlesworth et al 2022
From Mendel to quantitative genetics in the genome era: the scientific legacy of W. G. Hill
“Lithium Response in Bipolar Disorder: Genetics, Genomics, and Beyond”, Papiol et al 2022
Lithium response in bipolar disorder: Genetics, genomics, and beyond
“Systematic Comparison of Family History and Polygenic Risk across 24 Common Diseases”, Mars et al 2022
Systematic comparison of family history and polygenic risk across 24 common diseases
“Medical Genetics in the 19th Century As Background to the Development of Psychiatric Genetics”, Kendler 2022
Medical genetics in the 19th century as background to the development of psychiatric genetics
“Physical Activity and Health: Findings from Finnish Monozygotic Twin Pairs Discordant for Physical Activity”, Kujala et al 2022
“Examining Social Genetic Effects on Educational Attainment via Parental Educational Attainment, Income, and Parenting”, Su et al 2022
“Clarifying the Causes of Consistent and Inconsistent Findings in Genetics”, Dattani et al 2022
Clarifying the causes of consistent and inconsistent findings in genetics
“An Anatomy of the Intergenerational Correlation of Educational Attainment—Learning from the Educational Attainments of Norwegian Twins and Their Children”, Baier et al 2022
“Sex Differences in the Genetic and Environmental Underpinnings of Meat and Plant Preferences”, Çınar et al 2022
Sex differences in the genetic and environmental underpinnings of meat and plant preferences
“Secretoglobin Family 1D Member 2 (SCGB1D2) Protein Inhibits Growth of Borrelia Burgdorferi and Affects Susceptibility to Lyme Disease”, Strausz et al 2022
“How Full Is the Evolutionary Fuel Tank? A Meta-Analysis Quantifies the Heritable Genetic Variance in Fitness—The Fuel of Evolution”, Walsh 2022
“Human Brain Anatomy Reflects Separable Genetic and Environmental Components of Socioeconomic Status”, Kweon et al 2022
Human brain anatomy reflects separable genetic and environmental components of socioeconomic status
“Reweighting the UK Biobank to Reflect Its Underlying Sampling Population Substantially Reduces Pervasive Selection Bias due to Volunteering”, Alten et al 2022
“Fast and Accurate Bayesian Polygenic Risk Modeling With Variational Inference”, Zabad et al 2022
Fast and Accurate Bayesian Polygenic Risk Modeling with Variational Inference
“Evaluating Indirect Genetic Effects of Siblings Using Singletons”, Howe et al 2022
Evaluating indirect genetic effects of siblings using singletons
“Birth Order Differences in Education Originate in Postnatal Environments”, Isungset et al 2022
Birth order differences in education originate in postnatal environments
“Epistasis and Adaptation on Fitness Landscapes”, Bank 2022
“Genomics, Convergent Neuroscience and Progress in Understanding Autism Spectrum Disorder”, Willsey et al 2022
Genomics, convergent neuroscience and progress in understanding autism spectrum disorder
“Multi-Omics Analyses Cannot Identify True-Positive Novel Associations from Underpowered Genome-Wide Association Studies of Four Brain-Related Traits”, Baranger et al 2022
“Childhood Trauma and Borderline Personality Disorder Traits: A Discordant Twin Study”, Skaug et al 2022
Childhood trauma and borderline personality disorder traits: A discordant twin study
“Dominance versus Epistasis: the Biophysical Origins and Plasticity of Genetic Interactions within and between Alleles”, Xie et al 2022
“Genetic and Environmental Influences on Biological Essentialism, Heuristic Thinking, Need for Closure, and Conservative Values: Insights From a Survey and Twin Study”, Morosoli et al 2022
“New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias”, Bellenguez et al 2022
New insights into the genetic etiology of Alzheimer’s disease and related dementias
“Generalizability and Effect Measure Modification in Sibling Comparison Studies”, Sjölander et al 2022
Generalizability and effect measure modification in sibling comparison studies
“Sibling Comparison Studies”, Sjölander et al 2022
“Modeling Assortative Mating and Genetic Similarities between Partners, Siblings, and In-Laws”, Torvik et al 2022
Modeling assortative mating and genetic similarities between partners, siblings, and in-laws
“Multivariate Genomic Architecture of Cortical Thickness and Surface Area at Multiple Levels of Analysis”, Grotzinger et al 2022
“Quantifying Bias from Measurable & Unmeasurable Confounders Across 3 Domains of Individual Determinants of Political Preferences”, Ahlskog & Oskarsson 2022
“Significant Sparse Polygenic Risk Scores across 813 Traits in UK Biobank”, Tanigawa et al 2022
Significant sparse polygenic risk scores across 813 traits in UK Biobank
“Patterns of Item Nonresponse Behavior to Survey Questionnaires Are Systematic and Have a Genetic Basis”, Mignogna et al 2022
“The Genetics of Specific Cognitive Abilities”, Procopio et al 2022
“Measuring Biodiversity from DNA in the Air”, Clare et al 2022
“Discovery of Genomic Loci of the Human Cerebral Cortex Using Genetically Informed Brain Atlases”, Makowski et al 2022
Discovery of genomic loci of the human cerebral cortex using genetically informed brain atlases
“Idiosyncratic Learning Performance in Flies”, Smith et al 2022
“Multi-Ancestry EQTL Meta-Analysis of Human Brain Identifies Candidate Causal Variants for Brain-Related Traits”, Zeng et al 2022
“Alcohol Metabolism Genes and Risks of Site-Specific Cancers in Chinese Adults: An 11-Year Prospective Study”, Im et al 2022
“ExPRSweb—An Online Repository With Polygenic Risk Scores for Common Health-Related Exposures”, Ma et al 2022
ExPRSweb—An Online Repository with Polygenic Risk Scores for Common Health-related Exposures
“Interaction Testing and Polygenic Risk Scoring to Estimate the Association of Common Genetic Variants With Treatment Resistance in Schizophrenia”, Pardiñas et al 2022
“A Saturated Map of Common Genetic Variants Associated With Human Height from 5.4 Million Individuals of Diverse Ancestries”, Yengo et al 2022
“Identifying Imaging Genetic Associations via Regional Morphometricity Estimation”, Bao et al 2022
Identifying imaging genetic associations via regional morphometricity estimation
“Familial Resemblance, Citizenship, and Counterproductive Work Behavior: A Combined Twin, Adoption, Parent-Offspring, and Spouse Approach”, Anderson et al 2022b
“Role of Polygenic Risk Score in the Familial Transmission of Bipolar Disorder in Youth”, Birmaher et al 2021
Role of Polygenic Risk Score in the Familial Transmission of Bipolar Disorder in Youth
“Interest of Phenomic Prediction As an Alternative to Genomic Prediction in Grapevine”, Brault et al 2021
Interest of phenomic prediction as an alternative to genomic prediction in grapevine
“Evidence for Excess Familial Clustering of Post Traumatic Stress Disorder in the US Veterans Genealogy Resource”, Cannon-Albright et al 2021
“Multi-Ancestry Meta-Analysis of Asthma Identifies Novel Associations and Highlights the Value of Increased Power and Diversity”, Tsuo et al 2021
“Dietary Mediators of the Genetic Susceptibility to Obesity?Results from the Quebec Family Study”, Jacob et al 2021
Dietary Mediators of the Genetic Susceptibility to Obesity?Results from the Quebec Family Study
“Global Biobank Meta-Analysis Initiative: Powering Genetic Discovery across Human Diseases”, Initiative & Zhou 2021
Global Biobank Meta-analysis Initiative: powering genetic discovery across human diseases
“The Genetic Architecture of Obsessive-Compulsive Disorder: Contribution of Liability to OCD From Alleles Across the Frequency Spectrum”, Mahjani et al 2021
“Genetically Informed, Multilevel Analysis of the Flynn Effect across 4 Decades & 3 WISC Versions”, Giangrande et al 2021
Genetically informed, multilevel analysis of the Flynn Effect across 4 decades & 3 WISC versions
“Improving GWAS Discovery and Genomic Prediction Accuracy in Biobank Data”, Orliac et al 2021
Improving GWAS discovery and genomic prediction accuracy in Biobank data
“Returning Actionable Genomic Results in a Research Biobank: Analytic Validity, Clinical Implementation, and Resource Usage”, Zawatsky et al 2021
“Bridging the Explanatory Gaps: What Can We Learn from a Biological Agency Perspective?”, Sultan et al 2021
Bridging the explanatory gaps: What can we learn from a biological agency perspective?
“Parent-Of-Origin Effects in the UK Biobank”, Hofmeister et al 2021
“Genome-Wide Analysis of 53,400 People With Irritable Bowel Syndrome Highlights Shared Genetic Pathways With Mood and Anxiety Disorders”, Eijsbouts et al 2021
“Sex Impacts the Function of Major Depression-Linked Variants in Vivo”, Mulvey et al 2021
Sex impacts the function of major depression-linked variants in vivo
“The Genetic Basis of Spatial Cognitive Variation in a Food-Caching Bird”, Branch et al 2021
The genetic basis of spatial cognitive variation in a food-caching bird
“Predicting Skeletal Stature Using Ancient DNA”, Cox et al 2021
“Between-Group Mean Differences in Intelligence in the United States Are >0% Genetically Caused: Five Converging Lines of Evidence”, Warne 2021b
“Genomic Prediction in the Wild: A Case Study in Soay Sheep”, Ashraf et al 2021
“Polygenic Basis and Biomedical Consequences of Telomere Length Variation”, Codd et al 2021
Polygenic basis and biomedical consequences of telomere length variation
“Evolution of Sociability by Artificial Selection”, Scott et al 2021
“A Gene-Environment Interaction Study of Polygenic Scores and Maltreatment on Childhood ADHD”, He & Li 2021b
A Gene-Environment Interaction Study of Polygenic Scores and Maltreatment on Childhood ADHD
“Increased Somatic Mutation Burdens in Normal Human Cells due to Defective DNA Polymerases”, Robinson et al 2021
Increased somatic mutation burdens in normal human cells due to defective DNA polymerases
“Investigating Perceived Heritability of Mental Health Disorders and Attitudes toward Genetic Testing in the United States, United Kingdom, and Australia”, Morosoli et al 2021
“Pervasive Downward Bias in Estimates of Liability Scale Heritability in GWAS Meta-Analysis: A Simple Solution”, Grotzinger et al 2021
“The Debate Between Two of the Founders of American Psychiatric Genetics, Aaron Rosanoff and Abraham Myerson, on Mendelian Models for Psychiatric Illness (1911–1917)”, Kendler 2021b
“The Association between Polygenic Scores for Attention-Deficit/hyperactivity Disorder and School Performance: The Role of Attention-Deficit/hyperactivity Disorder Symptoms, Polygenic Scores for Educational Attainment, and Shared Familial Factors”, Jangmo et al 2021
“Genome-Wide Association Study of Musical Beat Synchronization Demonstrates High Polygenicity”, Niarchou et al 2021
Genome-wide association study of musical beat synchronization demonstrates high polygenicity
“Using Genes to Explore the Effects of Cognitive and Non-Cognitive Skills on Education and Labor Market Outcomes”, Buser et al 2021 (page 3)
“Discovery of 42 Genome-Wide Statistically-Significant Loci Associated With Dyslexia”, Doust et al 2021
Discovery of 42 Genome-Wide Statistically-Significant Loci Associated with Dyslexia
“Leveraging Fine-Mapping and Non-European Training Data to Improve Cross-Population Polygenic Risk Scores”, Weissbrod et al 2021
“Somatic Mutation Rates Scale With Lifespan across Mammals”, Cagan et al 2021
“The Omnigenic Model and Polygenic Prediction of Complex Traits”, Mathieson 2021
The omnigenic model and polygenic prediction of complex traits
“The Distribution of Common-Variant Effect Sizes”, O’Connor 2021
“Genome-Wide Methylation Data Improves Dissection of the Effect of Smoking on Body Mass Index”, Amador et al 2021
Genome-wide methylation data improves dissection of the effect of smoking on body mass index
“MegaLMM: Mega-Scale Linear Mixed Models for Genomic Predictions With Thousands of Traits”, Runcie et al 2021
MegaLMM: Mega-scale linear mixed models for genomic predictions with thousands of traits
“X-Chromosome Influences on Neuroanatomical Variation in Humans”, Mallard et al 2021b
X-chromosome influences on neuroanatomical variation in humans
“The Nature of Hereditary Influences on Insanity from Research on Asylum Records in Western Europe in the Mid-19th Century”, Kendler 2021
“Genome Scans of Facial Features in East Africans and Cross-Population Comparisons Reveal Novel Associations”, Liu et al 2021
“Genetics of Substance Use Disorders in the Era of Big Data”, Gelernter & Polimanti 2021
“The Chilean Socio-Ethno-Genomic Cline”, Barozet et al 2021
“Blood-Based Epigenome-Wide Analyses of Cognitive Abilities”, McCartney et al 2021
“Rapid Sequencing-Based Diagnosis of Thiamine Metabolism Dysfunction Syndrome”, Owen et al 2021
Rapid Sequencing-Based Diagnosis of Thiamine Metabolism Dysfunction Syndrome
“Genomic Characterization of World’s Longest Selection Experiment in Mouse Reveals the Complexity of Polygenic Traits”, Palma-Vera et al 2021
“Bi-Ancestral Depression GWAS in the Million Veteran Program and Meta-Analysis in >1.2 Million Individuals Highlight New Therapeutic Directions”, Levey et al 2021
“Loss-Of-Function Mutations in the Melanocortin 4 Receptor in a UK Birth Cohort”, Wade et al 2021
Loss-of-function mutations in the melanocortin 4 receptor in a UK birth cohort
“Genome-Wide Association Study of More Than 40,000 Bipolar Disorder Cases Provides New Insights into the Underlying Biology”, Mullins et al 2021
“Echolocating Bats Rely on an Innate Speed-Of-Sound Reference”, Amichai & Yovel 2021
Echolocating bats rely on an innate speed-of-sound reference
“Resource Profile and User Guide of the Polygenic Index Repository”, Becker et al 2021
Resource Profile and User Guide of the Polygenic Index Repository
“Multi-Scale Inference of Genetic Trait Architecture Using Biologically Annotated Neural Networks”, Demetci et al 2021
Multi-scale Inference of Genetic Trait Architecture using Biologically Annotated Neural Networks
“A Comparison of Ten Polygenic Score Methods for Psychiatric Disorders Applied across Multiple Cohorts”, Ni et al 2021
“Gene-Environment Correlations and Causal Effects of Childhood Maltreatment on Physical and Mental Health: a Genetically Informed Approach”, Warrier et al 2021
“Modification of Heritability for Educational Attainment and Fluid Intelligence by Socioeconomic Deprivation in the UK Biobank”, Rask-Andersen et al 2021
“Genetic and Environmental Sources of Familial Resemblance in Anxiety: a Nuclear Twin Family Design”, Ding et al 2021
Genetic and environmental sources of familial resemblance in anxiety: a nuclear twin family design
“An Expanded Set of Genome-Wide Association Studies of Brain Imaging Phenotypes in UK Biobank”, Smith et al 2021c
An expanded set of genome-wide association studies of brain imaging phenotypes in UK Biobank
“Are These Australia’s Most Identical Twins? Meet the Twinnies: Bridgette and Paula Powers”, News 2021
Are these Australia’s most identical twins? Meet the Twinnies: Bridgette and Paula Powers
“Toward a Fine-Scale Population Health Monitoring System”, Belbin et al 2021
“Genetic Attributions and Perceptions of Naturalness Are Shaped by Evaluative Valence”, Lebowitz et al 2021
Genetic attributions and perceptions of naturalness are shaped by evaluative valence
“Parental Characteristics and Offspring Mental Health and Related Outcomes: a Systematic Review of Genetically-Informative Literature”, Jami et al 2021
“Large Uncertainty in Individual PRS Estimation Impacts PRS-Based Risk Stratification”, Ding et al 2021
Large uncertainty in individual PRS estimation impacts PRS-based risk stratification
“Polygenic Risk for Depression, Anxiety and Neuroticism Are Associated With the Severity and Rate of Change in Depressive Symptoms across Adolescence”, Kwong et al 2021
“Evaluation of Polygenic Prediction Methodology within a Reference-Standardized Framework”, Pain et al 2021
Evaluation of polygenic prediction methodology within a reference-standardized framework
“High Trait Variability in Optimal Polygenic Prediction Strategy within Multiple-Ancestry Cohorts”, Lehmann et al 2021
High trait variability in optimal polygenic prediction strategy within multiple-ancestry cohorts
“Using DNA to Predict Intelligence”, Stumm & Plomin 2021
“GWAS in Almost 195,000 Individuals Identifies 50 Previously Unidentified Genetic Loci for Eye Color”, Simcoe et al 2021
GWAS in almost 195,000 individuals identifies 50 previously unidentified genetic loci for eye color
“Genes, Ideology, and Sophistication”, Kalmoe & Johnson 2021
“Therapygenetic Effects of 5-HTTLPR on Cognitive-Behavioral Therapy in Anxiety Disorders: A Meta-Analysis”, Schiele et al 2021
“Using DNA to Predict Behavior Problems from Preschool to Adulthood”, Gidziela et al 2021
Using DNA to predict behavior problems from preschool to adulthood
“Phenotypic Covariance across the Entire Spectrum of Relatedness for 86 Billion Pairs of Individuals”, Kemper et al 2021
Phenotypic covariance across the entire spectrum of relatedness for 86 billion pairs of individuals
“The Augmented Classical Twin Design: Incorporating Genome-Wide Identity by Descent Sharing Into Twin Studies in Order to Model Violations of the Equal Environments Assumption”, Hwang et al 2021
“Pathfinder: A Gamified Measure to Integrate General Cognitive Ability into the Biological, Medical and Behavioral Sciences”, Malanchini et al 2021
“Identification of 370 Genetic Loci for Age at First Sex and Birth Linked to Externalizing Behavior”, Mills et al 2021
Identification of 370 genetic loci for age at first sex and birth linked to externalizing behavior
“The Heritability of Reading and Reading-Related Neurocognitive Components: A Multi-Level Meta-Analysis”, Andreola et al 2021
“Genetics and Child Development: Recent Advances and Their Implications for Developmental Research”, Armstrong-Carter et al 2021
Genetics and Child Development: Recent Advances and Their Implications for Developmental Research
“Genetic and Environmental Influences of Dietary Indices in a UK Female Twin Cohort”, Mompeo et al 2021
Genetic and Environmental Influences of Dietary Indices in a UK Female Twin Cohort
“Developmental Trajectories of Delinquent and Aggressive Behavior: Evidence for Differential Heritability”, Isen et al 2021
“From Genotype to Phenotype: Polygenic Prediction of Complex Human Traits”, Raben et al 2021
From Genotype to Phenotype: polygenic prediction of complex human traits
“Nurture Might Be Nature: Cautionary Tales and Proposed Solutions”, Hart et al 2021
Nurture might be nature: cautionary tales and proposed solutions
“Differences between Germline Genomes of Monozygotic Twins”, Jonsson et al 2021
“Twin-Singleton Comparisons Across Multiple Domains of Life”, Willemsen et al 2021
“Dr Rona Strawbridge”
“The Genetic Architecture of Depression in Individuals of East Asian Ancestry: A Genome-Wide Association Study”, Giannakopoulou et al 2021
“Evidence of Horizontal Indirect Genetic Effects in Humans”, Xia et al 2020
“Does the Brain-Derived Neurotrophic Factor Val66Met Polymorphism Modulate the Effects of Physical Activity and Exercise on Cognition?”, Heras et al 2020
“10 Years of Enhancing Neuro-Imaging Genetics through Meta-Analysis: An Overview from the ENIGMA Genetics Working Group”, Medland et al 2020
“Largest GWAS (n = 1,126,563) of Alzheimer’s Disease Implicates Microglia and Immune Cells”, Wightman et al 2020
Largest GWAS (n = 1,126,563) of Alzheimer’s Disease Implicates Microglia and Immune Cells
“The Heritability of Insomnia: A Meta-Analysis of Twin Studies”, Barclay et al 2020
The heritability of insomnia: A meta-analysis of twin studies
“Twins Living Apart: Behavioral Insights/Twin Study Reviews: Managing Monochorionic-Diamniotic Twin Pregnancies; Paternity Testing in Multiple Pregnancies; Twin Research on Resilience; Trisomies in Twin Pregnancies/Human Interest: Reunited Brazilian Twins; Website for Twins With Disabled Co-Twins; Twins Separated in Secret of the Nile Series; Mengele: Unmasking the Angel of Death; Twins Helping Others”, Segal 2020
“An Integrative Analysis of Genomic and Exposomic Data for Complex Traits and Phenotypic Prediction”, Zhou & Lee 2020
An integrative analysis of genomic and exposomic data for complex traits and phenotypic prediction
“Estimation of Non-Additive Genetic Variance in Human Complex Traits from a Large Sample of Unrelated Individuals”, Hivert et al 2020
“‘Landmark’ Study Resolves a Major Mystery of How Genes Govern Human Height”, Kaiser 2020
‘Landmark’ study resolves a major mystery of how genes govern human height
“Individuals With Common Diseases but With a Low Polygenic Risk Score Could Be Prioritized for Rare Variant Screening”, Lu et al 2020c
“Could Polygenic Risk Scores Be Useful in Psychiatry? A Review”, Murray et al 2020
Could Polygenic Risk Scores Be Useful in Psychiatry? A Review
“Exploring the Variance in Complex Traits Captured by DNA Methylation Assays”, Battram et al 2020
Exploring the variance in complex traits captured by DNA methylation assays
“Happiness and Wellbeing; the Value and Findings from Genetic Studies”, Weijer et al 2020
Happiness and Wellbeing; the value and findings from genetic studies
“Genome-Wide Association Study of over 40,000 Bipolar Disorder Cases Provides Novel Biological Insights”, Mullins et al 2020
“Causes of Variation in Food Preference in the Netherlands”, Vink et al 2020
“Genetic Fortune: Winning or Losing Education, Income, and Health”, Kweon et al 2020 (page 4)
Genetic Fortune: Winning or Losing Education, Income, and Health
“Improved Genetic Prediction of Complex Traits from Individual-Level Data or Summary Statistics”, Zhang et al 2020
Improved genetic prediction of complex traits from individual-level data or summary statistics
“Sibling Validation of Polygenic Risk Scores and Complex Trait Prediction”, Lello et al 2020
Sibling validation of polygenic risk scores and complex trait prediction
“On the Genetic Basis of Political Orientation”, Daws & Weinschenk 2020
“Genetic and Environmental Variation in Educational Attainment: an Individual-Based Analysis of 28 Twin Cohorts”, Silventoinen et al 2020
“The Genes We Inherit and Those We Don’t: Maternal Genetic Nurture and Child BMI Trajectories”, Tubbs et al 2020
The Genes We Inherit and Those We Don’t: Maternal Genetic Nurture and Child BMI Trajectories
“Association of Parental Substance Misuse With Offspring Substance Misuse and Criminality: a Genetically Informed Register-Based Study”, Latvala et al 2020
“Recent Common Origin, Reduced Population Size, and Marked Admixture Have Shaped European Roma Genomes”, Bianco et al 2020
“Germline Mutation Rates in Young Adults Predict Longevity and Reproductive Lifespan”, Cawthon et al 2020
Germline mutation rates in young adults predict longevity and reproductive lifespan
“Efficient Polygenic Risk Scores for Biobank Scale Data by Exploiting Phenotypes from Inferred Relatives”, Truong et al 2020
“Combined Utility of 25 Disease and Risk Factor Polygenic Risk Scores for Stratifying Risk of All-Cause Mortality”, Meisner et al 2020
“Discovery of 318 New Risk Loci for Type 2 Diabetes and Related Vascular Outcomes among 1.4 Million Participants in a Multi-Ancestry Meta-Analysis”, Vujkovic et al 2020
“Preimplantation Genetic Testing for Polygenic Disease Relative Risk Reduction: Evaluation of Genomic Index Performance in 11,883 Adult Sibling Pairs”, Treff et al 2020
“Human Postprandial Responses to Food and Potential for Precision Nutrition”, Berry et al 2020
Human postprandial responses to food and potential for precision nutrition
“The Polygenic Architecture of Schizophrenia—Rethinking Pathogenesis and Nosology”, Smeland et al 2020
The polygenic architecture of schizophrenia—rethinking pathogenesis and nosology
“Risk in Relatives, Heritability, SNP-Based Heritability, and Genetic Correlations in Psychiatric Disorders: A Review”, Baselmans et al 2020
“Large-Scale Genome-Wide Association Study in a Japanese Population Identifies Novel Susceptibility Loci across Different Diseases”, Ishigaki et al 2020
“The Earliest Origins of Genetic Nurture: The Prenatal Environment Mediates the Association Between Maternal Genetics and Child Development”, Armstrong-Carter et al 2020
“A Fast and Scalable Framework for Large-Scale and Ultrahigh-Dimensional Sparse Regression With Application to the UK Biobank”, Qian et al 2020
“Exploring Genetic Contributions to News Use Motives and Frequency of News Consumption: A Study of Identical and Fraternal Twins”, York & Haridakis 2020
“Genetic Risk Scores for Cardiometabolic Traits in Sub-Saharan African Populations”, Ekoru et al 2020
Genetic Risk Scores for Cardiometabolic Traits in Sub-Saharan African Populations
“Genome-Wide Analysis Identifies Genetic Effects on Reproductive Success and Ongoing Natural Selection at the FADS Locus”, Mathieson et al 2020
“GWAS of Depression Phenotypes in the Million Veteran Program and Meta-Analysis in More Than 1.2 Million Participants Yields 178 Independent Risk Loci”, Levey et al 2020
“Musings on Visscher Et Al 2006”, Visscher 2020
“Insights into the Genetic Architecture of the Human Face”, White et al 2020
“Heritability of Affectionate Communication: A Twins Study”, Floyd et al 2020
“Using Genetics for Social Science”, Harden & Koellinger 2020
“Sociology, Genetics, and the Coming of Age of Sociogenomics”, Mills & Tropf 2020
“Beyond Culture and the Family: Evidence from Twin Studies on the Genetic and Environmental Contribution to Values”, Twito & Knafo-Noam 2020
“Greater Variability in Chimpanzee (Pan Troglodytes) Brain Structure among Males”, DeCasien et al 2020
Greater variability in chimpanzee (Pan troglodytes) brain structure among males
“Identifying Genetic Variants Underlying Phenotypic Variation in Plants without Complete Genomes”, Voichek & Weigel 2020
Identifying genetic variants underlying phenotypic variation in plants without complete genomes
“Birmingham and Beyond”, Eaves 2020
“Nicholas G. Martin and the Extended Twin Model”, Maes 2020b
“The SNP-Based Heritability—A Commentary on Yang Et Al 2010”, Yang 2020
“Genetic Endowments and Wealth Inequality”, Barth et al 2020
“The Barbarians Are at the Gate!”, Hatemi 2020
“Sociopolitical Attitudes Through the Lens of Behavioral Genetics: Contributions from Dr Nicholas Martin”, Verhulst 2020
“Functional Connectome-Wide Associations of Schizophrenia Polygenic Risk”, Cao et al 2020
Functional connectome-wide associations of schizophrenia polygenic risk
“The Multiplex Model of the Genetics of Alzheimer’s Disease”, Sims et al 2020
“The Behavioral, Cellular and Immune Mediators of HIV-1 Acquisition: New Insights from Population Genetics”, Powell et al 2020
“Analysis of Variance When Both Input and Output Sets Are High-Dimensional”, Campos et al 2020
Analysis of variance when both input and output sets are high-dimensional
“The Association of Oxytocin Receptor Gene (OXTR) Polymorphisms Antisocial Behavior: A Meta-Analysis”, Poore & Waldman 2020
The Association of Oxytocin Receptor Gene (OXTR) Polymorphisms Antisocial Behavior: A Meta-analysis
“Genetic Architecture of Complex Traits and Disease Risk Predictors”, Yong et al 2020
Genetic Architecture of Complex Traits and Disease Risk Predictors
“Using Human Genetics to Understand the Disease Impacts of Testosterone in Men and Women”, Ruth et al 2020
Using human genetics to understand the disease impacts of testosterone in men and women
“Studies of Human Twins Reveal Genetic Variation That Affects Dietary Fat Perception”, Lin et al 2020
Studies of human twins reveal genetic variation that affects dietary fat perception
“Theoretical and Empirical Quantification of the Accuracy of Polygenic Scores in Ancestry Divergent Populations”, Wang et al 2020
“Educational Attainment Polygenic Scores in Hungary: Evidence for Validity and a Historical Gene-Environment Interaction”, Ujma et al 2020
“Reproducible Genetic Risk Loci for Anxiety: Results From ∼200,000 Participants in the Million Veteran Program”, Levey et al 2020
“Heritability in Friendship Networks”, Neugart & Yildirim 2020
“A Brief History of Human Disease Genetics”, Claussnitzer et al 2020
“Multitrait Analysis of Glaucoma Identifies New Risk Loci and Enables Polygenic Prediction of Disease Susceptibility and Progression”, Craig 2020
“GWAS of 165,084 Japanese Individuals Identified 9 Loci Associated With Dietary Habits”, Matoba 2020
GWAS of 165,084 Japanese individuals identified 9 loci associated with dietary habits
“Introducing M-GCTA a Software Package to Estimate Maternal (Or Paternal) Genetic Effects on Offspring Phenotypes”, Qiao 2020
“Obesity and Eating Behavior from the Perspective of Twin and Genetic Research”, Silventoinen 2020
Obesity and eating behavior from the perspective of twin and genetic research
“How Genetic and Environmental Variance in Personality Traits Shift across the Life Span: Evidence from a Cross-National Twin Study”, Kandler et al 2020
“Genome-Wide Analysis Identifies Molecular Systems and 149 Genetic Loci Associated With Income”, Hill et al 2019
Genome-wide analysis identifies molecular systems and 149 genetic loci associated with income
“Beyond SNP Heritability: Polygenicity and Discoverability of Phenotypes Estimated With a Univariate Gaussian Mixture Model”, Holland et al 2019
“Multivariable G-E Interplay in the Prediction of Educational Achievement”, Allegrini et al 2019
Multivariable G-E interplay in the prediction of educational achievement
“The Louisville Twin Study: Past, Present and Future”, Davis et al 2019
“Genome-Wide Association Studies in Schizophrenia: Recent Advances, Challenges and Future Perspective”, Dennison et al 2019
Genome-wide association studies in schizophrenia: Recent advances, challenges and future perspective
“Predicting Educational Achievement from Genomic Measures and Socioeconomic Status”, Stumm et al 2019
Predicting educational achievement from genomic measures and socioeconomic status
“Genes, Gender Inequality, and Educational Attainment”, Herd et al 2019
“Population-Specific and Transethnic Genome-Wide Analyses Reveal Distinct and Shared Genetic Risks of Coronary Artery Disease”, Koyama et al 2019
“Structural Variation in the Sequencing Era”, Ho et al 2019
“The Propensity for Aggressive Behavior and Lifetime Incarceration Risk: A Test for Gene-Environment Interaction (G × E) Using Whole-Genome Data”, Barnes et al 2019
“Uganda Genome Resource Enables Insights into Population History and Genomic Discovery in Africa”, Gurdasani et al 2019b
Uganda Genome Resource Enables Insights into Population History and Genomic Discovery in Africa
“Associations of Autozygosity With a Broad Range of Human Phenotypes”, Clark et al 2019
Associations of autozygosity with a broad range of human phenotypes
“Sibling Comparisons Elucidate the Associations between Educational Attainment Polygenic Scores and Alcohol, Nicotine and Cannabis”, Salvatore et al 2019
“Genome-Wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations”, Peterson et al 2019
“Genomics of Human Aggression: Current State of Genome-Wide Studies and an Automated Systematic Review Tool”, Odintsova et al 2019
“Polygenic Score Analysis Of Educational Achievement And Intergenerational Mobility”, Rustichini et al 2019
Polygenic Score Analysis Of Educational Achievement And Intergenerational Mobility
“Genetic ‘General Intelligence’, Objectively Determined and Measured”, Fuente et al 2019
Genetic ‘General Intelligence’, Objectively Determined and Measured
“Software As a Service for the Genomic Prediction of Complex Diseases”, Bolli et al 2019
Software as a Service for the Genomic Prediction of Complex Diseases
“In-Field Whole Plant Maize Architecture Characterized by Latent Space Phenotyping”, Gage et al 2019
In-field whole plant maize architecture characterized by Latent Space Phenotyping
“Children of the Twins Early Development Study (CoTEDS): A Children-Of-Twins Study”, Ahmadzadeh et al 2019
Children of the Twins Early Development Study (CoTEDS): A Children-of-Twins Study
“The Study of Personality Architecture and Dynamics (SPeADy): A Longitudinal and Extended Twin Family Study”, Kandler et al 2019c
“Mendelian Gene Discovery: Fast and Furious With No End in Sight”, Bamshad et al 2019
Mendelian Gene Discovery: Fast and Furious with No End in Sight
“Dumb or Smart Asses? Donkey’s (Equus Asinus) Cognitive Capabilities Share the Heritability and Variation Patterns of Human’s (Homo Sapiens) Cognitive Capabilities”, González et al 2019 (page 2)
“A Prospective Analysis of Genetic Variants Associated With Human Lifespan”, Wright et al 2019
A Prospective Analysis of Genetic Variants Associated with Human Lifespan
“The Higher Power of Religiosity Over Personality on Political Ideology”, Ksiazkiewicz & Friesen 2019
The Higher Power of Religiosity Over Personality on Political Ideology
“Leveraging European Infrastructures to Access 1 Million Human Genomes by 2022”, Saunders et al 2019
Leveraging European infrastructures to access 1 million human genomes by 2022
“Polygenic and Clinical Risk Scores and Their Impact on Age at Onset of Cardiometabolic Diseases and Common Cancers”, Mars et al 2019
“Genetic Architecture of Socioeconomic Outcomes: Educational Attainment, Occupational Status, and Wealth”, Liu 2019b
“A Mother’s Legacy: the Strength of Maternal Effects in Animal Populations”, Moore et al 2019
A mother’s legacy: the strength of maternal effects in animal populations
“An AI Expert’s Toughest Project: Writing Code to save His Son’s Life”
An AI expert’s toughest project: writing code to save his son’s life
“The Chinese National Twin Registry: a ‘Gold Mine’ for Scientific Research”, Gao et al 2019
The Chinese National Twin Registry: a ‘gold mine’ for scientific research
“Genomic Risk Score Offers Predictive Performance Comparable to Clinical Risk Factors for Ischaemic Stroke”, Abraham et al 2019
“Inferring the Nature of Missing Heritability in Human Traits Using Data from the GWAS Catalog”, López-Cortegano & Caballero 2019
Inferring the Nature of Missing Heritability in Human Traits Using Data from the GWAS Catalog
“Unravelling the Genetic Basis of Schizophrenia and Bipolar Disorder With GWAS: A Systematic Review”, Prata et al 2019
Unravelling the genetic basis of schizophrenia and bipolar disorder with GWAS: A systematic review
“Gene Regulation and the Architecture of Complex Human Traits in the Genomics Era”, Boutwell & White 2019
Gene regulation and the architecture of complex human traits in the genomics era
“Genetic Influences on Antisocial Behavior: Recent Advances and Future Directions”, Gard et al 2019
Genetic influences on antisocial behavior: recent advances and future directions
“Making the Most of Clumping and Thresholding for Polygenic Scores”, Privé et al 2019
Making the most of Clumping and Thresholding for polygenic scores
“Breed Differences of Heritable Behavior Traits in Cats”, Salonen et al 2019
“Heritability of Lifetime Earnings”, Hyytinen et al 2019
“Genetic Variation across the Human Olfactory Receptor Repertoire Alters Odor Perception”, Trimmer et al 2019
Genetic variation across the human olfactory receptor repertoire alters odor perception
“Amid Animal Cruelty Debate, 80% of South Korea’s Sniffer Dogs Are Cloned”, Tribune 2019
Amid animal cruelty debate, 80% of South Korea’s sniffer dogs are cloned
“Supplement to No Support for Historic Candidate Gene or Candidate Gene-By-Interaction Hypotheses for Major Depression across Multiple Large Samples”, Border et al 2019
“Maternal and Fetal Genetic Effects on Birth Weight and Their Relevance to Cardio-Metabolic Risk Factors”, Warrington et al 2019
“The Heritability of Self-Control: A Meta-Analysis”, Willems et al 2019
“Is Population Structure in the Genetic Biobank Era Irrelevant, a Challenge, or an Opportunity?”, Lawson et al 2019
Is population structure in the genetic biobank era irrelevant, a challenge, or an opportunity?
“Is Apostasy Heritable? A Behavior Genetics Study”, Freeman 2019
“Latent Space Phenotyping: Automatic Image-Based Phenotyping for Treatment Studies”, Ubbens et al 2019
Latent Space Phenotyping: Automatic Image-Based Phenotyping for Treatment Studies
“A Major Role for Common Genetic Variation in Anxiety Disorders”, Purves et al 2019
A Major Role for Common Genetic Variation in Anxiety Disorders
“Simulation of Model Overfit in Variance Explained With Genetic Data”, Derringer 2019
Simulation of model overfit in variance explained with genetic data
“Comparing Within-Family and Between-Family Polygenic Score Prediction”, Selzam et al 2019
Comparing within-family and between-family polygenic score prediction
“Uncovering the Genetic Architecture of Major Depression”, McIntosh et al 2019
“Ant Collective Behavior Is Heritable and Shaped by Selection”, Walsh et al 2019
Ant collective behavior is heritable and shaped by selection
“Evidence That the Association of Childhood Trauma With Psychosis and Related Psychopathology Is Not Explained by Gene-Environment Correlation: A Monozygotic Twin Differences Approach”, Lecei et al 2019
“Molecular Support for Heterogonesis Resulting in Sesquizygotic Twinning”, Gabbett et al 2019
Molecular Support for Heterogonesis Resulting in Sesquizygotic Twinning
“Fast and Flexible Linear Mixed Models for Genome-Wide Genetics”, Runcie & Crawford 2019
Fast and flexible linear mixed models for genome-wide genetics
“Repurposing Large Health Insurance Claims Data to Estimate Genetic and Environmental Contributions in 560 Phenotypes”, Lakhani et al 2019
“Genome-Wide Meta-Analysis of Depression Identifies 102 Independent Variants and Highlights the Importance of the Prefrontal Brain Regions”, Howard et al 2019
“Adaptive Phenotypic Plasticity for Life-History and Less Fitness-Related Traits”, Acasuso-Rivero et al 2019
Adaptive phenotypic plasticity for life-history and less fitness-related traits
“Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort”, Bai et al 2019
Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort
“Polygenic Scores: a Public Health Hazard?”, Baverstock 2019
“Mitochondrial Alterations May Underlie Race-Specific Differences in Cancer Risk and Outcome”, Beebe-Dimmer & Cooney 2019
Mitochondrial alterations may underlie race-specific differences in cancer risk and outcome
“Testing for Family Influences on Obesity: The Role of Genetic Nurture”, Cawley et al 2019
Testing for family influences on obesity: The role of genetic nurture
“Family Networks versus Genetics in Social Outcomes, England 1750–2019 [Slides]”, Clark 2019
Family Networks versus Genetics in Social Outcomes, England 1750–2019 [slides]
“The Causes and Consequences of Genetic Interactions (Epistasis)”, Domingo et al 2019
The Causes and Consequences of Genetic Interactions (Epistasis)
“How Genome-Wide Association Studies (GWAS) Made Traditional Candidate Gene Studies Obsolete”, Duncan et al 2019
How genome-wide association studies (GWAS) made traditional candidate gene studies obsolete
“Genetic Variation, Comparative Genomics, and the Diagnosis of Disease”, Eichler 2019
Genetic Variation, Comparative Genomics, and the Diagnosis of Disease
“Identifying Facial Phenotypes of Genetic Disorders Using Deep Learning”, Gurovich et al 2019
Identifying facial phenotypes of genetic disorders using deep learning
“Genetic and Phenotypic Landscape of the Major Histocompatibilty Complex Region in the Japanese Population”, Hirata et al 2019
“New Insight into Human Sweet Taste: a Genome-Wide Association Study of the Perception and Intake of Sweet Substances”, Hwang et al 2019
“Comment on ‘Polygenic Scores: A Public Health Hazard?’”, Joyner 2019b
“Native American Groups Wary of Big U.S. Biobank: Tribes Say Health Officials Were Slow to Consult Them”
“Towards Clinical Utility of Polygenic Risk Scores”, Lambert et al 2019
“The Genetics of Human Skin and Hair Pigmentation”, Pavan & Sturm 2019
“Absolute and Relative Estimates of Genetic and Environmental Variance in Brain Structure Volumes”, Strike et al 2019
Absolute and relative estimates of genetic and environmental variance in brain structure volumes
“Benefits and Limitations of Genome-Wide Association Studies”, Tam et al 2019
“Correlations between Relatives: From Mendelian Theory to Complete Genome Sequence”, Thompson 2019
Correlations between relatives: From Mendelian theory to complete genome sequence
“Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine”, Udler et al 2019
Genetic risk scores for diabetes diagnosis and precision medicine
“From R. A. Fisher's 1918 Paper to GWAS a Century Later”, Visscher & Goddard 2019
“Free Will, Determinism, and Intuitive Judgments About the Heritability of Behavior”, Willoughby et al 2019b
Free Will, Determinism, and Intuitive Judgments About the Heritability of Behavior
“Complex Trait Prediction from Genome Data: Contrasting EBV in Livestock to PRS in Humans”, Wray et al 2019
Complex Trait Prediction from Genome Data: Contrasting EBV in Livestock to PRS in Humans
“The Dynamic Associations Between Cortical Thickness and General Intelligence Are Genetically Mediated”, Schmitt et al 2019
“Phenotypic Annotation: Using Polygenic Scores to Translate Discoveries From Genome-Wide Association Studies From the Top Down”, Belsky & Harden 2019
“Viewing Education Policy through a Genetic Lens”, Asbury & Wai 2019
“No Support for Historical Candidate Gene or Candidate Gene-By-Interaction Hypotheses for Major Depression Across Multiple Large Samples”, Border et al 2019
“Extreme Morning Chronotypes Are Often Familial and Not Exceedingly Rare: the Estimated Prevalence of Advanced Sleep Phase, Familial Advanced Sleep Phase, and Advanced Sleep-Wake Phase Disorder in a Sleep Clinic Population”, Curtis et al 2019
“The Nature and Nurture of HEXACO Personality Trait Differences: An Extended Twin Family Study”, Kandler et al 2019b
The nature and nurture of HEXACO personality trait differences: An extended twin family study
“A Genome-Wide Association Study of Bitter and Sweet Beverage Consumption”, Zhong et al 2019
A genome-wide association study of bitter and sweet beverage consumption
“Asymmetrical Genetic Attributions for Prosocial versus Antisocial Behavior”, Lebowitz et al 2019
Asymmetrical genetic attributions for prosocial versus antisocial behavior
“Genomic Prediction of Complex Disease Risk”, Lello et al 2018
“Variation in the Heritability of Child Body Mass Index by Obesogenic Home Environment”, Schrempft et al 2018
Variation in the Heritability of Child Body Mass Index by Obesogenic Home Environment
“Genetic Consequences of Social Stratification in Great Britain”, Abdellaoui et al 2018
Genetic Consequences of Social Stratification in Great Britain
“Existence and Implications of Population Variance Structure”, Musharoff et al 2018
“Evidence for Bias of Genetic Ancestry in Resting State Functional MRI”, Altmann & Mourao-Miranda 2018
Evidence for Bias of Genetic Ancestry in Resting State Functional MRI
“A Twin Study on the Correlates of Voluntary Exercise Behavior in Adolescence”, Schutte et al 2018
A twin study on the correlates of voluntary exercise behavior in adolescence
“Polygenicity of Complex Traits Is Explained by Negative Selection”, O’Connor et al 2018
Polygenicity of complex traits is explained by negative selection
“Genomic Prediction of Cognitive Traits in Childhood and Adolescence”, Allegrini et al 2018
Genomic prediction of cognitive traits in childhood and adolescence
“Meta-Analysis of Genome-Wide Association Studies for Body Fat Distribution in 694,649 Individuals of European Ancestry”, Pulit et al 2018
“Evidence for Gene-Environment Correlation in Child Feeding: Links between Common Genetic Variation for BMI in Children and Parental Feeding Practices”, Selzam et al 2018
“Genes, Education, and Labor Market Outcomes: Evidence from the Health and Retirement Study”, Papageorge & Thom 2018
Genes, Education, and Labor Market Outcomes: Evidence from the Health and Retirement Study
“Genome-Wide Statistically-Significant Regions in 43 Utah High-Risk Families Implicate Multiple Genes Involved in Risk for Completed Suicide”, Coon et al 2018
“The Genetics of the Mood Disorder Spectrum: Genome-Wide Association Analyses of over 185,000 Cases and 439,000 Controls”, Coleman et al 2018
“On the Genetic and Environmental Sources of Social and Political Participation in Adolescence and Early Adulthood”, Kornadt et al 2018
“Modeling Functional Enrichment Improves Polygenic Prediction Accuracy in UK Biobank and 23andMe Data Sets”, Márquez-Luna et al 2018
“Genomic Underpinnings of Lifespan Allow Prediction and Reveal Basis in Modern Risks”, Timmers et al 2018
Genomic underpinnings of lifespan allow prediction and reveal basis in modern risks
“Multi-Ethnic Genome-Wide Association Study for Atrial Fibrillation”, Roselli et al 2018
Multi-ethnic genome-wide association study for atrial fibrillation
“Study of 300,486 Individuals Identifies 148 Independent Genetic Loci Influencing General Cognitive Function”, Davies et al 2018
“German Law Allows Use of DNA to Predict Suspects' Looks”, Vogel 2018
“The Genetic Architecture of Hair Color in the UK Population”, Morgan et al 2018
“A Multimethodological Study of Preschoolers’ Preferences for Aggressive Television and Video Games”, Jamnik & DiLalla 2018
A Multimethodological Study of Preschoolers’ Preferences for Aggressive Television and Video Games
“Genome-Wide Association Study of Social Genetic Effects on 170 Phenotypes in Laboratory Mice”, Baud et al 2018
Genome-wide association study of social genetic effects on 170 phenotypes in laboratory mice
“On the Genetic and Genomic Basis of Aggression, Violence, and Antisocial Behavior”, Beaver et al 2018
On the Genetic and Genomic Basis of Aggression, Violence, and Antisocial Behavior
“Novel Susceptibility Loci and Genetic Regulation Mechanisms for Type 2 Diabetes”, Xue et al 2018
Novel susceptibility loci and genetic regulation mechanisms for type 2 diabetes
“Better Estimation of SNP Heritability from Summary Statistics Provides a New Understanding of the Genetic Architecture of Complex Traits”, Speed & Balding 2018
“The Nature of Nurture: Using a Virtual-Parent Design to Test Parenting Effects on Children’s Educational Attainment in Genotyped Families”, Bates et al 2018
“Quantitative Analysis of Population-Scale Family Trees With Millions of Relatives”, Kaplanis et al 2018
Quantitative analysis of population-scale family trees with millions of relatives
“Meta-Analysis of Genome-Wide Association Studies for Cattle Stature Identifies Common Genes That Regulate Body Size in Mammals”, Bouwman et al 2018
“Genome-Wide Study Identifies 611 Loci Associated With Risk Tolerance and Risky Behaviors”, Linnér et al 2018
Genome-wide study identifies 611 loci associated with risk tolerance and risky behaviors
“The Genomic Commons”, Contreras & Knoppers 2018
“Genomic Risk Prediction of Coronary Artery Disease in Nearly 500,000 Adults: Implications for Early Screening and Primary Prevention”, Inouye et al 2018
“Genetic Architecture of Gene Expression Traits across Diverse Populations”, Mogil et al 2018
Genetic architecture of gene expression traits across diverse populations
“Fine-Mapping of an Expanded Set of Type 2 Diabetes Loci to Single-Variant Resolution Using High-Density Imputation and Islet-Specific Epigenome Maps”, Mahajan et al 2018
“Mixed Model Association for Biobank-Scale Data Sets”, Loh et al 2018
“Genome-Wide Association Study of 1 Million People Identifies 111 Loci for Atrial Fibrillation”, Nielsen et al 2018
Genome-wide association study of 1 million people identifies 111 loci for atrial fibrillation
“Generalizing Genetic Risk Scores from Europeans to Hispanics/Latinos”, Grinde et al 2018
Generalizing Genetic Risk Scores from Europeans to Hispanics/Latinos
“The Shared Genetic Basis of Human Fluid Intelligence and Brain Morphology”, Ge et al 2018
The Shared Genetic Basis of Human Fluid Intelligence and Brain Morphology
“A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease”, Abul-Husn et al 2018
A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease
“Muscle Health and Performance in Monozygotic Twins With 30 Years of Discordant Exercise Habits”, Bathgate 2018
Muscle health and performance in monozygotic twins with 30 years of discordant exercise habits
“Joint Contributions of Rare Copy Number Variants and Common SNPs to Risk for Schizophrenia”, Bergen et al 2018
Joint Contributions of Rare Copy Number Variants and Common SNPs to Risk for Schizophrenia
“No Association between Urbanisation, Neighbourhood Deprivation and IBD: a Population-Based Study of 4 Million Individuals”
“Runs of Homozygosity: Windows into Population History and Trait Architecture”, Ceballos et al 2018
Runs of homozygosity: windows into population history and trait architecture
“Genome-Wide Mapping of Global-To-Local Genetic Effects on Human Facial Shape”, Claes et al 2018
Genome-wide mapping of global-to-local genetic effects on human facial shape
“Genotype Imputation from Large Reference Panels”, Das et al 2018
“The Arrival of Social Science Genomics”, Freese 2018
“Maternal Prenatal Depressive Symptoms and Risk for Early-Life Psychopathology in Offspring: Genetic Analyses in the Norwegian Mother and Child Birth Cohort Study”, Hannigan et al 2018
“A Decade in Psychiatric GWAS Research”, Horwitz et al 2018
“Genome-Wide Association Meta-Analysis of Individuals of European Ancestry Identifies New Loci Explaining a Substantial Fraction of Hair Color Variation and Heritability”, Hysi et al 2018
“Exploring the Relationship between Polygenic Risk for Cannabis Use, Peer Cannabis Use and the Longitudinal Course of Cannabis Involvement”, Johnson et al 2018c
“Genome-Wide Polygenic Scores for Common Diseases Identify Individuals With Risk Equivalent to Monogenic Mutations”, Khera et al 2018
“Genetics of Blood Lipids among ~300,000 Multi-Ethnic Participants of the Million Veteran Program”, Klarin et al 2018
Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program
“The Impact of Variation in Twin Relatedness on Estimates of Heritability and Environmental Influences”, Liu 2018
“‘The Fixity of Whiteness’: Genetic Admixture and the Legacy of the One-Drop Rule”, Liz 2018
‘The Fixity of Whiteness’: Genetic Admixture and the Legacy of the One-Drop Rule
“Refining the Accuracy of Validated Target Identification through Coding Variant Fine-Mapping in Type 2 Diabetes”, Mahajan et al 2018
“Predicting Polygenic Risk of Psychiatric Disorders”, Martin et al 2018b
“Genetic Influences on Musical Specialization: a Twin Study on Choice of Instrument and Music Genre”
Genetic influences on musical specialization: a twin study on choice of instrument and music genre
“Inferring Transmission Histories of Rare Alleles in Population-Scale Genealogies”, Nelson et al 2018
Inferring Transmission Histories of Rare Alleles in Population-Scale Genealogies
“Relationship between Deleterious Variation, Genomic Autozygosity, and Disease Risk: Insights from The 1000 Genomes Project”, Pemberton & Szpiech 2018
“The Biological Contributions to Gender Identity and Gender Diversity: Bringing Data to the Table”, Polderman 2018
The Biological Contributions to Gender Identity and Gender Diversity: Bringing Data to the Table
“Genetic Influence on Social Outcomes during and After the Soviet Era in Estonia”, Rimfeld et al 2018
Genetic influence on social outcomes during and after the Soviet era in Estonia
“Pairs of Genetically Unrelated Look-Alikes”, Segal et al 2018
“GWAS for BMI: a Treasure Trove of Fundamental Insights into the Genetic Basis of Obesity”, Speakman et al 2018
GWAS for BMI: a treasure trove of fundamental insights into the genetic basis of obesity
“Genome-Wide Association Meta-Analysis Highlights Light-Induced Signaling As a Driver for Refractive Error”, Tedja et al 2018
“Developmental Origins of Chronic Physical Aggression: A Bio-Psycho-Social Model for the Next Generation of Preventive Interventions”, Tremblay et al 2018
“Convergence of Placenta Biology and Genetic Risk for Schizophrenia”, Ursini et al 2018
Convergence of placenta biology and genetic risk for schizophrenia
“Genetic Variation in Health Insurance Coverage”, Wehby & Shane 2018
“Fine-Mapping and Functional Studies Highlight Potential Causal Variants for Rheumatoid Arthritis and Type 1 Diabetes”, Westra et al 2018
“A Differential Evolution Approach to Feature Selection in Genomic Prediction”, Whalen 2018
A Differential Evolution Approach to Feature Selection in Genomic Prediction
“Twin Classroom Dilemma: To Study Together or Separately?”
“Identifying Loci Affecting Trait Variability and Detecting Interactions in Genome-Wide Association Studies”, Young et al 2018
“Unravelling Quasi-Causal Environmental Effects via Phenotypic and Genetically Informed Multi-Rater Models: The Case of Differential Parenting and Authoritarianism”, Zapko-Willmes et al 2018
“Genome-Wide Analyses Using UK Biobank Data Provide Insights into the Genetic Architecture of Osteoarthritis”, Zengini et al 2018
“Estimation of Complex Effect-Size Distributions Using Summary-Level Statistics from Genome-Wide Association Studies across 32 Complex Traits”, Zhang et al 2018c
“The Genetics of Fruit Flavour Preferences”, Klee & Tieman 2018
“The Personal and Clinical Utility of Polygenic Risk Scores”, Torkamani et al 2018
“Ideology Between the Lines”, Hannikainen 2018
“Social and Genetic Pathways in Multigenerational Transmission of Educational Attainment”, Liu 2018b
Social and Genetic Pathways in Multigenerational Transmission of Educational Attainment
“Personality and Genetic Associations With Military Service”, Miles & Haider-Markel 2018
“Sizing up Whole-Genome Sequencing Studies of Common Diseases”, Wray 2018b
Sizing up whole-genome sequencing studies of common diseases
“The New Genetics of Intelligence”, Plomin & Stumm 2018
“86 Genomic Sites Associated With Educational Attainment Provide Insight into the Biology of Cognitive Performance”, Lee 2018
“Misestimation of Heritability and Prediction Accuracy of Male-Pattern Baldness”, Yap 2018
Misestimation of heritability and prediction accuracy of male-pattern baldness
“Revisiting the Children-Of-Twins Design: Improving Existing Models for the Exploration of Intergenerational Associations”, McAdams et al 2018
“Relatedness Disequilibrium Regression Estimates Heritability without Environmental Bias”, Young et al 2018
Relatedness disequilibrium regression estimates heritability without environmental bias
“The Genetics of University Success”, Smith-Woolley et al 2018
“DeepGS: Predicting Phenotypes from Genotypes Using Deep Learning”, Ma et al 2017
DeepGS: Predicting phenotypes from genotypes using Deep Learning
“The Nature of Nurture: Effects of Parental Genotypes”, Kong et al 2017
“Estimating Heritability without Environmental Bias”, Young et al 2017
“Implicit Causal Models for Genome-Wide Association Studies”, Tran & Blei 2017
“Socioeconomic Status and Genetic Influences on Cognitive Development”, Figlioa et al 2017
Socioeconomic status and genetic influences on cognitive development
“Genome-Wide Meta-Analysis Associates HLA-DQA1/DRB1 and LPA and Lifestyle Factors With Human Longevity”, Joshi et al 2017
“Genetic Analysis of over One Million People Identifies 535 Novel Loci for Blood Pressure”, Evangelou et al 2017
Genetic analysis of over one million people identifies 535 novel loci for blood pressure
“Biological Insights Into Muscular Strength: Genetic Findings in the UK Biobank”, Tikkanen et al 2017
Biological Insights Into Muscular Strength: Genetic Findings in the UK Biobank
“Accurate Genomic Prediction Of Human Height”, Lello et al 2017
“Multiethnic Meta-Analysis Identifies New Loci for Pulmonary Function”, Wyss et al 2017
Multiethnic meta-analysis identifies new loci for pulmonary function
“Genetic Diversity Turns a New PAGE in Our Understanding of Complex Traits”, Wojcik et al 2017
Genetic Diversity Turns a New PAGE in Our Understanding of Complex Traits
“Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register”, Hilker et al 2017
“Genome-Wide Association Study of Habitual Physical Activity in over 277,000 UK Biobank Participants Identifies Novel Variants and Genetic Correlations With Chronotype and Obesity-Related Traits”, Klimentidis et al 2017
“The Genetic Basis of Human Brain Structure and Function: 1,262 Genome-Wide Associations Found from 3,144 GWAS of Multimodal Brain Imaging Phenotypes from 9,707 UK Biobank Participants”, Elliott et al 2017
“Identifying Genetic Variants That Affect Viability in Large Cohorts”, Mostafavi et al 2017
Identifying genetic variants that affect viability in large cohorts
“Genome-Wide Genetic Data on ~500,000 UK Biobank Participants”, Bycroft et al 2017
Genome-wide genetic data on ~500,000 UK Biobank participants
“Statistical Correction of the Winner’s Curse Explains Replication Variability in Quantitative Trait Genome-Wide Association Studies”, Palmer & Pe’er 2017
“10 Years of GWAS Discovery: Biology, Function, and Translation”, Visscher et al 2017
10 Years of GWAS Discovery: Biology, Function, and Translation
“Sexual Dimorphism in the Genetic Influence on Human Childlessness”, Verweij et al 2017
Sexual dimorphism in the genetic influence on human childlessness
“Targeting Aggression in Severe Mental Illness: The Predictive Role of Genetic, Epigenetic, and Metabolomic Markers”, Manchia & Fanos 2017
“Discovery of the First Genome-Wide Statistically-Large Risk Loci for ADHD”, Demontis et al 2017
Discovery of the first genome-wide statistically-large risk loci for ADHD
“Codebreaker: A Deeply Personal Quest Made Matthew Might a Leader in Precision Medicine and Brought Him to UAB”
“Genetic Predisposition to Obesity and Medicare Expenditures”, Wehby et al 2017
“Father Absence and Accelerated Reproductive Development”, Gaydosh et al 2017
“Comparison of Methods That Use Whole Genome Data to Estimate the Heritability and Genetic Architecture of Complex Traits”, Evans et al 2017
“Psychiatric Genomics: An Update and an Agenda”, Sullivan et al 2017
“Biogeographic Ancestry and Socioeconomic Outcomes in the Americas: A Meta-Analysis”, Kirkegaard et al 2017
Biogeographic Ancestry and Socioeconomic Outcomes in the Americas: A Meta-Analysis
“Analysis of Genetic Similarity among Friends and Schoolmates in the National Longitudinal Study of Adolescent to Adult Health (Add Health)”, Domingue et al 2017
“Personalized Media: A Genetically-Informative Investigation of Individual Differences in Online Media Use”, Ayorech et al 2017
“Genetic and Environmental Influences on Household Financial Distress”, Xu et al 2017
Genetic and Environmental Influences on Household Financial Distress
“Covariate Selection for Association Screening in Multiphenotype Genetic Studies”, Aschard et al 2017b
Covariate selection for association screening in multiphenotype genetic studies
“A Genetic Basis of Economic Egalitarianism”, Batricevic & Littvay 2017
“Copy Number Variation in Syndromic Forms of Psychiatric Illness: The Emerging Value of Clinical Genetic Testing in Psychiatry”
“Co-Aggregation of Major Psychiatric Disorders in Individuals With First-Degree Relatives With Schizophrenia: a Nationwide Population-Based Study”, Cheng et al 2017
“Predictive Accuracy of Combined Genetic and Environmental Risk Scores”
Predictive accuracy of combined genetic and environmental risk scores
“Little Evidence That Socioeconomic Status Modifies Heritability of Literacy and Numeracy in Australia”
“Internet Addiction and Its Facets: The Role of Genetics and the Relation to Self-Directedness”, Hahn et al 2017
Internet addiction and its facets: The role of genetics and the relation to self-directedness
“National Clinical Audit Data Decodes the Genetic Architecture of Developmental Dysplasia of the Hip”, Hatzikotoulas et al 2017
National clinical audit data decodes the genetic architecture of developmental dysplasia of the hip
“Effect of Sequence Variants on Variance in Glucose Levels Predicts Type 2 Diabetes Risk and Accounts for Heritability”, Ivarsdottir et al 2017
“Inference on the Genetic Basis of Eye and Skin Color in an Admixed Population via Bayesian Linear Mixed Models”
“Polygenic Scores via Penalized Regression on Summary Statistics”
Polygenic scores via penalized regression on summary statistics
“Differences in Genetic and Environmental Variation in Adult Body Mass Index by Sex, Age, Time Period, and Region: an Individual-Based Pooled Analysis of 40 Twin Cohorts”
“Protein-Altering Variants Associated With Body Mass Index Implicate Pathways That Control Energy Intake and Expenditure in Obesity”, Turcot et al 2017
“2017 Constance Holden Memorial Address: Liberal Creationism”
“Polygenic Prediction of Obsessive Compulsive Symptoms”, Zilhão et al 2017
“Racial Minority Group Interest in Direct-To-Consumer Genetic Testing: Findings from the PGen Study”, Landry et al 2017
Racial minority group interest in direct-to-consumer genetic testing: findings from the PGen study
“Beyond Questionable Research Methods: The Role of Omitted Relevant Research in the Credibility of Research”, Schmidt 2017
“Twin Spouses and Unrelated Look-Alikes: New Views”, Segal 2017
“Heritability of Working in a Creative Profession”, Roeling et al 2017
“The Heritability of Autism Spectrum Disorder”, Sandin et al 2017
“Associations of Coffee Genetic Risk Scores With Coffee, Tea and Other Beverages in the UK Biobank”, Taylor & Munafò 2016
Associations of coffee genetic risk scores with coffee, tea and other beverages in the UK Biobank
“Genetic Variation in the Social Environment Contributes to Health and Disease”, Baud et al 2016
Genetic Variation in the Social Environment Contributes to Health and Disease
“Statistical Properties of Simple Random-Effects Models for Genetic Heritability”, Steinsaltz et al 2016
Statistical properties of simple random-effects models for genetic heritability
“Genome-Wide Meta-Analysis of Cognitive Empathy: Heritability, and Correlates With Sex, Neuropsychiatric Conditions and Brain Anatomy”, Warrier et al 2016
“Evidence That Lower Socioeconomic Position Accentuates Genetic Susceptibility to Obesity”, Tyrrell et al 2016
Evidence that lower socioeconomic position accentuates genetic susceptibility to obesity
“Genome-Wide Association Study of Antisocial Personality Disorder”, Rautiainen et al 2016
Genome-wide association study of antisocial personality disorder
“Genetic Prediction of Male Pattern Baldness”, Hagenaars et al 2016
“How Cognitive Genetic Factors Influence Fertility Outcomes: A Mediational SEM Analysis”, Menie et al 2016
How cognitive genetic factors influence fertility outcomes: A mediational SEM analysis
“Phenome-Wide Heritability Analysis of the UK Biobank”, Ge et al 2016
“Estimate of Disease Heritability Using 4.7 Million Familial Relationships Inferred from Electronic Health Records”, Polubriaginof et al 2016
“Genome-Wide Variants of Eurasian Facial Shape Differentiation and DNA Based Face Prediction”, Qiao et al 2016
Genome-wide Variants of Eurasian Facial Shape Differentiation and DNA based Face Prediction
“Cost-Effectiveness of Pharmacogenetic-Guided Treatment: Are We There Yet?”, Verbelen et al 2016
Cost-effectiveness of pharmacogenetic-guided treatment: are we there yet?
“Genome-Wide Association Study Reveals Multiple Loci Influencing Normal Human Facial Morphology”, Shaffer et al 2016
Genome-Wide Association Study Reveals Multiple Loci Influencing Normal Human Facial Morphology
“Genome-Wide Association Study Identifies 74 Loci Associated With Educational Attainment”, Okbay et al 2016
Genome-wide association study identifies 74 loci associated with educational attainment
“Mega-Analysis of 31,396 Individuals from 6 Countries Uncovers Strong Gene-Environment Interaction for Human Fertility”, Tropf et al 2016
“The Great Migration and African-American Genomic Diversity”, Baharian et al 2016
“Mortality Selection in a Genetic Sample and Implications for Association Studies”, Domingue et al 2016
Mortality Selection in a Genetic Sample and Implications for Association Studies
“Genome-Wide Estimates of Heritability for Social Demographic Outcomes”, Domingue et al 2016
Genome-Wide Estimates of Heritability for Social Demographic Outcomes
“Molecular Genetic Contributions to Social Deprivation and Household Income in UK Biobank (n = 112,151)”, Hill et al 2016
“Older Fathers’ Children Have Lower Evolutionary Fitness across Four Centuries and in Four Populations”, Arslan et al 2016
“How Does a Cowbird Learn To Be a Cowbird? New Research Explains How These Brood Parasites—Who Are Raised by Other Species—Still Manage to Become Cowbirds”, Soniak 2016
“Estimating Effect Sizes and Expected Replication Probabilities from GWAS Summary Statistics”, Holland et al 2016
Estimating Effect Sizes and Expected Replication Probabilities from GWAS Summary Statistics
“The Evolutionary Genetics of Personality Revisited”, Penke & Jokela 2016
“Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data”, Shi et al 2016
Contrasting the genetic architecture of 30 complex traits from summary association data
“Paraphilic Sexual Interests and Sexually Coercive Behavior: A Population-Based Twin Study”, Baur et al 2016
Paraphilic Sexual Interests and Sexually Coercive Behavior: A Population-Based Twin Study
“Genome-Wide Association Study of Working Memory Brain Activation”, Blokland et al 2016
Genome-wide association study of working memory brain activation
“Developing and Evaluating Polygenic Risk Prediction Models for Stratified Disease Prevention”, Chatterjee et al 2016
Developing and evaluating polygenic risk prediction models for stratified disease prevention
“Familial Aggregation of Attention-Deficit/hyperactivity Disorder”, Chen et al 2016
Familial aggregation of attention-deficit/hyperactivity disorder
“Genetic and Environmental Parent–Child Transmission of Value Orientations: An Extended Twin Family Study”
“The MC1R Gene and Youthful Looks”, Liu et al 2016
“Heritability and Causal Reasoning”, Lynch 2016
“A Genetic Epidemiological Mega Analysis of Smoking Initiation in Adolescents”, Murugadoss 2016
A Genetic Epidemiological Mega Analysis of Smoking Initiation in Adolescents
“School Achievement and Risk of Eating Disorders in a Swedish National Cohort”, Sundquist et al 2016
School Achievement and Risk of Eating Disorders in a Swedish National Cohort
“GWAS for Executive Function and Processing Speed Suggests Involvement of the CADM2 Gene”, Ibrahim-Verbaas et al 2016
GWAS for executive function and processing speed suggests involvement of the CADM2 gene
“PHENIX: A Multiple-Phenotype Imputation Method for Genetic Studies”, Dahl et al 2016
PHENIX: A multiple-phenotype imputation method for genetic studies
“A Novel Sibling-Based Design to Quantify Genetic and Shared Environmental Effects: Application to Drug Abuse, Alcohol Use Disorder and Criminal Behavior”, Kendler et al 2016
“Chorionicity and Heritability Estimates from Twin Studies: The Prenatal Environment of Twins and Their Resemblance Across a Large Number of Traits”, Beijsterveldt et al 2016
“Genetic and Environmental Influences on Food Preferences in Adolescence”, Smith et al 2016
Genetic and environmental influences on food preferences in adolescence
“Out on Their Own: a Test of Adult-Assisted Dispersal in Fledgling Brood Parasites Reveals Solitary Departures from Hosts”, Louder et al 2015
“Heritability of Neuroanatomical Shape”, Ge et al 2015
“An Empirical Bayes Mixture Model for Effect Size Distributions in Genome-Wide Association Studies”, Thompson et al 2015
An Empirical Bayes Mixture Model for Effect Size Distributions in Genome-Wide Association Studies
“Individual Esthetic Preferences for Faces Are Shaped Mostly by Environments, Not Genes”, Germine et al 2015
Individual esthetic Preferences for Faces Are Shaped Mostly by Environments, Not Genes
“Meta-Analysis of Twin Studies Highlights the Importance of Genetic Variation in Primary School Educational Achievement”, Zeeuw et al 2015
“SNP Hits on Cognitive Ability from 300k Individuals”, Hsu 2015
“Genetic Variance Estimation With Imputed Variants Finds Negligible Missing Heritability for Human Height and Body Mass Index”, Yang et al 2015
“Haplotypes of Common SNPs Can Explain Missing Heritability of Complex Diseases”, Bhatia et al 2015
Haplotypes of common SNPs can explain missing heritability of complex diseases
“The Support of Human Genetic Evidence for Approved Drug Indications”, Tipney et al 2015
The support of human genetic evidence for approved drug indications
“Signaling and Productivity in the Private Financial Returns to Schooling”, Bingley et al 2015
Signaling and Productivity in the Private Financial Returns to Schooling
“Meta-Analysis of the Heritability of Human Traits Based on 50 Years of Twin Studies”, Polderman et al 2015
Meta-analysis of the heritability of human traits based on 50 years of twin studies
“The Genetic Architecture of Pediatric Cognitive Abilities in the Philadelphia Neurodevelopmental Cohort”, Robinson et al 2015
“Genetic and Environmental Influences on Obesity-Related Phenotypes in Chinese Twins Reared Apart and Together”, Zhou et al 2015
“Is the Effect of Parental Education on Offspring Biased or Moderated by Genotype?”, Conley et al 2015
Is the Effect of Parental Education on Offspring Biased or Moderated by Genotype?
“Genetic Contributions to Variation in General Cognitive Function: a Meta-Analysis of Genome-Wide Association Studies in the CHARGE Consortium (N = 53,949)”, Davies et al 2015
“Is That Me or My Twin? Lack of Self-Face Recognition Advantage in Identical Twins”, Martini et al 2015
Is That Me or My Twin? Lack of Self-Face Recognition Advantage in Identical Twins
“The Contribution of Additive Genetic Variation to Personality Variation: Heritability of Personality”, Dochtermann et al 2015
The contribution of additive genetic variation to personality variation: heritability of personality
“HERITABILITY STUDIES IN THE POSTGENOMIC ERA: THE FATAL FLAW IS CONCEPTUAL*”
HERITABILITY STUDIES IN THE POSTGENOMIC ERA: THE FATAL FLAW IS CONCEPTUAL*
“Personality Related Traits As Predictors of Music Practice: Underlying Environmental and Genetic Influences”, Butkovic et al 2015
“Heritability of Compulsive Internet Use in Adolescents”, Vink et al 2015
“Physical Activity, Fitness, Glucose Homeostasis, and Brain Morphology in Twins”, Rottensteiner et al 2015
Physical activity, fitness, glucose homeostasis, and brain morphology in twins
“Genetic Studies of Body Mass Index Yield New Insights for Obesity Biology”, Locke et al 2015
Genetic studies of body mass index yield new insights for obesity biology
“Genome-Wide Meta-Analysis Identifies Six Novel Loci Associated With Habitual Coffee Consumption”, Cornelis et al 2015
Genome-wide meta-analysis identifies six novel loci associated with habitual coffee consumption
“Novel Loci Associated With Usual Sleep Duration: the CHARGE Consortium Genome-Wide Association Study”, Gottlieb et al 2015
Novel loci associated with usual sleep duration: the CHARGE Consortium Genome-Wide Association Study
“Sexual Offending Runs in Families: A 37-Year Nationwide Study”, Långström et al 2015
Sexual offending runs in families: A 37-year nationwide study
“LD Score Regression Distinguishes Confounding from Polygenicity in Genome-Wide Association Studies”, Bulik-Sullivan et al 2015
LD Score regression distinguishes confounding from polygenicity in genome-wide association studies
“LDpred: Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores”, Vilhjálmsson et al 2015
LDpred: Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores
“The Fine-Scale Genetic Structure of the British Population”, Leslie et al 2015
“Multi-Ancestry Genome-Wide Association Study of 21,000 Cases and 95,000 Controls Identifies New Risk Loci for Atopic Dermatitis”, Paternoster et al 2015
“Genetic Background of Extreme Violent Behavior”, Tiihonen et al 2015
“Association Analyses Identify 38 Susceptibility Loci for Inflammatory Bowel Disease and Highlight Shared Genetic Risk across Populations”, Liu et al 2015
“Genome-Wide Identification of MicroRNAs Regulating Cholesterol and Triglyceride Homeostasis”, Wagschal 2015
Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis
“Common Polygenic Variation Enhances Risk Prediction for Alzheimer’s Disease”, Escott-Price et al 2015
Common polygenic variation enhances risk prediction for Alzheimer’s disease
“The Intergenerational Transmission of Anxiety: A Children-Of-Twins Study”, Eley et al 2015
The Intergenerational Transmission of Anxiety: A Children-of-Twins Study
“Shared Genetic Influences Between Attention-Deficit/Hyperactivity Disorder (ADHD) Traits in Children and Clinical ADHD”, Stergiakouli 2015
“The Etiologic Role of Genetic and Environmental Factors in Criminal Behavior As Determined from Full & Half-Sibling Pairs: an Evaluation of the Validity of the Twin Method”, Kendler et al 2014
“Political Attitudes Develop Independently of Personality Traits”, Hatemi & Verhulst 2014
Political Attitudes Develop Independently of Personality Traits
“Simultaneous Discovery, Estimation and Prediction Analysis of Complex Traits Using a Bayesian Mixture Model”, Moser et al 2014
“Attention-Deficit/Hyperactivity Disorder Polygenic Risk Scores Predict Attention Problems in a Population-Based Sample of Children”, Groen-Blokhuis et al 2014
“On the Genetic Architecture of Intelligence and Other Quantitative Traits”, Hsu 2014
On the genetic architecture of intelligence and other quantitative traits
“Biological Insights from 108 Schizophrenia-Associated Genetic Loci”, Consortium 2014
Biological insights from 108 schizophrenia-associated genetic loci
“Nature, Nurture, and Expertise”, Plomin et al 2014
“Genetic Variation Associated With Differential Educational Attainment in Adults Has Anticipated Associations With School Performance in Children”, Ward et al 2014
“Heritability of Creative Achievement”, Piffer & Hur 2014
“Results of a ‘GWAS Plus:’ General Cognitive Ability Is Substantially Heritable and Massively Polygenic”, Kirkpatrick et al 2014
“Pulling Back The Curtain On Heritability Studies: Biosocial Criminology In The Postgenomic Era”, Burt & Simons 2014
Pulling Back The Curtain On Heritability Studies: Biosocial Criminology In The Postgenomic Era
“Genome-Wide Association Study of Proneness to Anger”, Mick et al 2014
“Genetic and Environmental Influences on Problematic Internet Use: A Twin Study”, Deryakulu & Ursavaş 2014
Genetic and environmental influences on problematic Internet use: A twin study
“Social Influence Constrained by the Heritability of Attitudes”, Schwab 2014
Social influence constrained by the heritability of attitudes
“Uncovering the Hidden Risk Architecture of the Schizophrenias: Confirmation in 3 Independent Genome-Wide Association Studies”, Arnedo 2014
“The Genetics of Music Accomplishment: Evidence for Gene-Environment Correlation and Interaction”, Hambrick & Tucker-Drob 2014
The genetics of music accomplishment: Evidence for gene-environment correlation and interaction
“Understanding the Relative Contributions of Direct Environmental Effects and Passive Genotype-Environment Correlations in the Association between Familial Risk Factors and Child Disruptive Behavior Disorders”, Bornovalova et al 2014
“Genetics of Rheumatoid Arthritis Contributes to Biology and Drug Discovery”, Okada et al 2014
Genetics of rheumatoid arthritis contributes to biology and drug discovery
“Genome-Wide Trans-Ancestry Meta-Analysis Provides Insight into the Genetic Architecture of Type 2 Diabetes Susceptibility”, Mahajan et al 2014
“The Genetic Basis of Music Ability”, Tan et al 2014
“Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies 6 New Risk Loci for Parkinson’s Disease”, Nalls et al 2014
“Parent-Of-Origin-Specific Allelic Associations among 106 Genomic Loci for Age at Menarche”, Perry et al 2014
Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche
“Defining the Role of Common Variation in the Genomic and Biological Architecture of Adult Human Height”, Wood et al 2014
“A Genome-Wide Association Study of Anorexia Nervosa”, Boraska et al 2014
“Replicability and Robustness of Genome-Wide-Association Studies for Behavioral Traits”, Rietveld et al 2014
Replicability and robustness of genome-wide-association studies for behavioral traits
“Impact of Measurement Error on Testing Genetic Association With Quantitative Traits”, Liao et al 2013
Impact of Measurement Error on Testing Genetic Association with Quantitative Traits
“Heritability and the Equal Environments Assumption: Evidence from Multiple Samples of Misclassified Twins”, Conley et al 2013
“GWAS of 126,559 Individuals Identifies Genetic Variants Associated With Educational Attainment”, Rietveld et al 2013
GWAS of 126,559 Individuals Identifies Genetic Variants Associated with Educational Attainment
“Using Extended Genealogy to Estimate Components of Heritability for 23 Quantitative and Dichotomous Traits”, Zaitlen et al 2013
“Childhood Intelligence Is Heritable, Highly Polygenic and Associated With FNBP1L”, Benyamin et al 2013
Childhood intelligence is heritable, highly polygenic and associated with FNBP1L
“Genetic Architecture of Skin and Eye Color in an African-European Admixed Population”, Beleza et al 2013
Genetic Architecture of Skin and Eye Color in an African-European Admixed Population
“An Opposite-Direction Modulation of the COMT Val158Met Polymorphism on the Clinical Response to Intrathecal Morphine and Triptans”, Cargnin et al 2013
“Common DNA Variants Predict Tall Stature in Europeans”, Liu et al 2013
“Contribution of Common Genetic Variants to Antidepressant Response”, Tansey et al 2013
Contribution of Common Genetic Variants to Antidepressant Response
“Should Evolutionary Geneticists Worry about Higher-Order Epistasis?”, Weinreich et al 2013
Should evolutionary geneticists worry about higher-order epistasis?
“Unrelated Look-Alikes: Replicated Study of Personality Similarity and Qualitative Findings on Social Relatedness”
“Personality Similarity in Unrelated Look-Alike Pairs: Addressing a Twin Study Challenge”
Personality similarity in unrelated look-alike pairs: Addressing a twin study challenge
“The Wilson Effect: The Increase in Heritability of IQ With Age”
The Wilson Effect: The Increase in Heritability of IQ With Age
“Http://www.science.sciencemag.org/highwire/filestream/594571/field_highwire_adjunct_files/1/Rietveld.SM.revision.2.pdf”
“The Swedish Twin Registry: Establishment of a Biobank and Other Recent Developments”, Magnusson 2013
The Swedish Twin Registry: establishment of a biobank and other recent developments
“Genetic Risk Factors for BMI and Obesity in an Ethnically Diverse Population: Results from the Population Architecture Using Genomics and Epidemiology (PAGE) Study”, Fesinmeyer et al 2013
“Tests of a Direct Effect of Childhood Abuse on Adult Borderline Personality Disorder Traits: a Longitudinal Discordant Twin Design”, Bornovalova et al 2013
“Common DNA Markers Can Account for More Than Half of the Genetic Influence on Cognitive Abilities”, Plomin et al 2013
Common DNA markers can account for more than half of the genetic influence on cognitive abilities
“Large-Scale Association Analysis Identifies New Risk Loci for Coronary Artery Disease”, Deloukas et al 2013
Large-scale association analysis identifies new risk loci for coronary artery disease
“A Meta-Analysis Identifies New Loci Associated With Body Mass Index in Individuals of African Ancestry”, Monda et al 2013
“Critical Need for Family-Based, Quasi-Experimental Designs in Integrating Genetic and Social Science Research”, D’Onofrio et al 2013
“A Genome-Wide Association Study of Behavioral Disinhibition”, McGue et al 2013
“Genome-Wide Meta-Analysis Identifies 11 New Loci for Anthropometric Traits and Provides Insights into Genetic Architecture”, Berndt et al 2013
“Large-Scale Genotyping Identifies a New Locus at 22q13.2 Associated With Female Breast Size”, Li et al 2013
Large-scale genotyping identifies a new locus at 22q13.2 associated with female breast size
“The Half-Life of DNA in Bone: Measuring Decay Kinetics in 158 Dated Fossils”, Allentoft et al 2012
The half-life of DNA in bone: measuring decay kinetics in 158 dated fossils
“Genetics of Aggression”, Anholt & Mackay 2012
“Unraveling the Genetic Etiology of Adult Antisocial Behavior: A Genome-Wide Association Study”, Tielbeek et al 2012
Unraveling the Genetic Etiology of Adult Antisocial Behavior: A Genome-Wide Association Study
“The Continuing Value of Twin Studies in the Omics Era”, Dongen et al 2012
“Correlation and Causation in the Study of Personality”, Lee 2012
“Genetic and Environmental Influences on Media Use and Communication Behaviors”, Kirzinger et al 2012
Genetic and Environmental Influences on Media Use and Communication Behaviors
“Sex Differences in Educational Attainment”, Mikk et al 2012
“The Heritability and Genetic Correlates of Mobile Phone Use: a Twin Study of Consumer Behavior”, Miller et al 2012
The heritability and genetic correlates of mobile phone use: a twin study of consumer behavior
“Fullerton Virtual Twin Study: An Update”, Segal et al 2012
“Relationship between Adiposity and Admixture in African-American and Hispanic-American Women”, Nassir et al 2012
Relationship between adiposity and admixture in African-American and Hispanic-American women
“Five Years of GWAS Discovery”, Visscher et al 2012
“Large-Scale Association Analysis Provides Insights into the Genetic Architecture and Pathophysiology of Type 2 Diabetes”, Morris et al 2012
“Effect of Shared Environmental Factors on Exercise Behavior from Age 7 to 12 Years”, Huppertz et al 2012
Effect of shared environmental factors on exercise behavior from age 7 to 12 years
“The Heritability of Avoidant and Dependent Personality Disorder Assessed by Personal Interview and Questionnaire”, Gjerde et al 2012
“Most Reported Genetic Associations With General Intelligence Are Probably False Positives”, Chabris et al 2012
Most reported genetic associations with general intelligence are probably false positives
“Genetic Variants and Associations of 25-Hydroxyvitamin D Concentrations With Major Clinical Outcomes”, Levin et al 2012
Genetic variants and associations of 25-hydroxyvitamin D concentrations with major clinical outcomes
“Research in China on the Molecular Genetics of Schizophrenia”, Cui & Jiang 2012
Research in China on the molecular genetics of schizophrenia
“Genetic and Environmental Influences on Individual Differences in Frequency of Play With Pets among Middle-Aged Men: A Behavioral Genetic Analysis”, Jacobson et al 2012
“Genomic Ancestry, Self-Reported ‘Color’ and Quantitative Measures of Skin Pigmentation in Brazilian Admixed Siblings”, Leite et al 2011
“Reconsidering the Heritability of Intelligence in Adulthood: Taking Assortative Mating and Cultural Transmission into Account”, Vinkhuyzen et al 2011
“A Novel, Functional and Replicable Risk Gene Region for Alcohol Dependence Identified by Genome-Wide Association Study”, Zuo et al 2011
“Large-Scale Genome-Wide Association Analysis of Bipolar Disorder Identifies a New Susceptibility Locus near ODZ4”, Sklar et al 2011
“Genome-Wide Association Studies Establish That Human Intelligence Is Highly Heritable and Polygenic”, Davies et al 2011
Genome-wide association studies establish that human intelligence is highly heritable and polygenic
“The Story of Emily and Control”, Alexander 2011
“The Biological Roots of Complex Thinking: Are Heritable Attitudes More Complex?”, Conway et al 2011
The Biological Roots of Complex Thinking: Are Heritable Attitudes More Complex?
“Exercise Participation in Adolescents and Their Parents: Evidence for Genetic and Generation Specific Environmental Effects”, Moor et al 2011
“Variation in Actual Relationship As a Consequence of Mendelian Sampling and Linkage”, Hill & Weir 2011
Variation in actual relationship as a consequence of Mendelian sampling and linkage
“Genome-Wide Association Study of the Child Behavior Checklist Dysregulation Profile”, Mick et al 2011
Genome-wide association study of the child behavior checklist dysregulation profile
“Alcohol Dependence in Men: Reliability and Heritability”, Ystrom et al 2011
“A Genome-Wide Association Study of Aging”, Walter et al 2011
“A Critical Review of the First 10 Years of Candidate Gene-By-Environment Interaction Research in Psychiatry”, Duncan & Keller 2011
“Meta-Analysis of Genome-Wide Association Studies of Asthma in Ethnically Diverse North American Populations”, Torgerson et al 2011
“Genome-Wide Association Study of Conduct Disorder Symptomatology”, Dick et al 2011
Genome-wide association study of conduct disorder symptomatology
“Estimating the Total Number of Susceptibility Variants Underlying Complex Diseases from Genome-Wide Association Studies”, So et al 2010
“On the Heritability of Consumer Decision Making: An Exploratory Approach for Studying Genetic Effects on Judgment and Choice”, Simonson & Sela 2010
“Consistent Association of Type 2 Diabetes Risk Variants Found in Europeans in Diverse Racial and Ethnic Groups”, Waters et al 2010
“In Search of Genes Associated With Risk for Psychopathic Tendencies in Children: a Two-Stage Genome-Wide Association Study of Pooled DNA”, Viding et al 2010
“Not by Twins Alone: Using the Extended Family Design to Investigate Genetic Influence on Political Beliefs”, Hatemi et al 2010
“Environmental Influences on Children’s Physical Activity: Quantitative Estimates Using a Twin Design”, Fisher et al 2010
Environmental Influences on Children’s Physical Activity: Quantitative Estimates Using a Twin Design
“A Longitudinal Study on Genetic and Environmental Influences on Leisure Time Physical Activity in the Finnish Twin Cohort”, Aaltonen et al 2010
“No Effect of Classroom Sharing on Educational Achievement in Twins: a Prospective, Longitudinal Cohort Study”, Polderman et al 2010
“Admixture Mapping Comes of Age”, Winkler et al 2010
“Human Face Recognition Ability Is Specific and Highly Heritable”, Wilmer et al 2010
Human face recognition ability is specific and highly heritable
“On Epistasis: Why It Is Unimportant in Polygenic Directional Selection”, Crow 2010
On epistasis: why it is unimportant in polygenic directional selection
“Genetic and Environmental Influences on the Transmission of Parental Depression to Children’s Depression and Conduct Disturbance: an Extended Children of Twins Study”, Silberg et al 2010
“Effects of Modafinil on the Sleep EEG Depend on Val158Met Genotype of COMT”, Bodenmann & Landolt 2010
Effects of modafinil on the sleep EEG depend on Val158Met genotype of COMT
“Meta-Analysis of Genome-Wide Association Studies of Attention-Deficit/hyperactivity Disorder”, Neale et al 2010
Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder
“Blood Vitamin D Levels in Relation to Genetic Estimation of African Ancestry”, Signorello et al 2010
Blood vitamin d levels in relation to genetic estimation of African ancestry
“Hundreds of Variants Clustered in Genomic Loci and Biological Pathways Affect Human Height”, Allen et al 2010
Hundreds of variants clustered in genomic loci and biological pathways affect human height
“Common SNPs Explain a Large Proportion of the Heritability for Human Height”, Yang et al 2010
Common SNPs explain a large proportion of the heritability for human height
“Genetic Ancestry, Social Classification, and Racial Inequalities in Blood Pressure in Southeastern Puerto Rico”, Gravlee et al 2009
“Genetics of Human Aggressive Behavior”, Craig & Halton 2009
“The Heritability of General Cognitive Ability Increases Linearly from Childhood to Young Adulthood”, Haworth et al 2009
The heritability of general cognitive ability increases linearly from childhood to young adulthood
“Musical Aptitude Is Associated With AVPR1A-Haplotypes”, Ukkola et al 2009
“Factors Contributing to the Facial Aging of Identical Twins”, Guyuron et al 2009
“Material Values Are Largely in the Family: A Twin Study of Genetic and Environmental Contributions to Materialism”, Giddens et al 2009
“History of Behavior Genetics”, Loehlin 2009
“Genetics of Exercise Behavior”, Stubbe & Geus 2009
“Nature versus Nurture Is Nonsense: On the Necessity of an Integrated Genetic, Social, Developmental, and Personality Psychology”, Barlow 2009
“Association of Substance Use Disorders With Childhood Trauma but Not African Genetic Heritage in an African American Cohort”, Ducci et al 2009
“Parental Alcoholism and Offspring Behavior Problems: Findings in Australian Children of Twins”, Waldron et al 2009
Parental alcoholism and offspring behavior problems: findings in Australian children of twins
“Sexy Sons and Sexy Daughters: the Influence of Parents’ Facial Characteristics on Offspring”, Cornwell & Perrett 2008
Sexy sons and sexy daughters: the influence of parents’ facial characteristics on offspring
“Unique Environmental Effects on Physical Activity Participation: A Twin Study”, Duncan et al 2008
Unique Environmental Effects on Physical Activity Participation: A Twin Study
“Transmission of Attitudes Toward Abortion and Gay Rights: Effects of Genes, Social Learning and Mate Selection”, Eaves & Hatemi 2008b
“Heritability in the Genomics Era—Concepts and Misconceptions”, Visscher et al 2008
Heritability in the genomics era—concepts and misconceptions
“Data and Theory Point to Mainly Additive Genetic Variance for Complex Traits”, Hill et al 2008
Data and Theory Point to Mainly Additive Genetic Variance for Complex Traits
“A Panel of Ancestry Informative Markers for Estimating Individual Biogeographical Ancestry and Admixture from 4 Continents: Utility and Applications”
“A Twin-Family Study of General IQ”, Leeuwen et al 2008
“Genetic Mapping in Human Disease”, Altshuler et al 2008
“Origins of Magic: Review of Genetic and Epigenetic Effects”, Ramagopalan et al 2007
“The Genetics of Political Attitudes”, Hatemi 2007
“A Children of Twins Study of Parental Divorce and Offspring Psychopathology”, D’Onofrio et al 2007
A Children of Twins Study of parental divorce and offspring psychopathology
“A Common Variant of HMGA2 Is Associated With Adult and Childhood Height in the General Population”, Weedon et al 2007
A common variant of HMGA2 is associated with adult and childhood height in the general population
“Genome Partitioning of Genetic Variation for Height from 11,214 Sibling Pairs”, Visscher 2007
Genome Partitioning of Genetic Variation for Height from 11,214 Sibling Pairs
“The Evolution of Personality Variation in Humans and Other Animals”, Nettle 2006
The evolution of personality variation in humans and other animals
“Achievement and Ascription in Educational Attainment: Genetic and Environmental Influences on Adolescent Schooling”, Nielsen 2006
“Genetic and Cultural Transmission of Smoking Initiation: An Extended Twin Kinship Model”, Maes et al 2006
Genetic and Cultural Transmission of Smoking Initiation: An Extended Twin Kinship Model
“Two Monozygotic Twin Pairs Discordant for Female-To-Male Transsexualism”, Segal 2006
Two Monozygotic Twin Pairs Discordant for Female-to-Male Transsexualism
“Smarter Than The Average Bug”, McCrone 2006
“Psychological Essentialism and Stereotype Endorsement”, Bastian & Haslam 2006
“Assumption-Free Estimation of Heritability from Genome-Wide Identity-By-Descent Sharing between Full Siblings”, Visscher et al 2006
“Influence of Environmental Factors on Facial Aging”, Rexbye et al 2006
“Genetic Determinism”, Anonymous 2006
“Harry Potter and the Recessive Allele”, Craig et al 2005
“Nature and Nurture: Genetic and Environmental Influences on Behavior”, Plomin & Asbury 2005b
Nature and Nurture: Genetic and Environmental Influences on Behavior
“Sports Participation during Adolescence: A Shift from Environmental to Genetic Factors”, Stubbe et al 2005
Sports Participation during Adolescence: A Shift from Environmental to Genetic Factors
“Personality Traits As Intermediary Phenotypes in Suicidal Behavior: Genetic Issues”, Baud 2005
Personality traits as intermediary phenotypes in suicidal behavior: Genetic issues
“Contributions of Parental Alcoholism, Prenatal Substance Exposure, and Genetic Transmission to Child ADHD Risk: A Female Twin Study”, Knopik et al 2005
“Genetic and Environmental Influences on Antisocial Behaviors: Evidence from Behavioral-Genetic Research”, Moffitt 2005
“Under the Skin: On the Impartial Treatment of Genetic and Environmental Hypotheses of Racial Differences”, Rowe 2005
“Personality Development: Stability and Change”, Caspi 2005
“The Cognitive Cost of Being a Twin: Evidence from Comparisons within Families in the Aberdeen Children of the 1950s Cohort Study”, Ronalds 2005
“Neurobiology of Intelligence: Science and Ethics”, Gray & Thompson 2004
“Intelligence: Genetics, Genes, and Genomics”, Plomin & Spinath 2004
“Genetic and Environmental Effects on Offspring Alcoholism: New Insights Using an Offspring-Of-Twins Design”, Jacob et al 2003
“Genetics and Educational Psychology”
“A Model-Fitting Implementation of the DeFries-Fulker Model for Selected Twin Data”
A Model-Fitting Implementation of the DeFries-Fulker Model for Selected Twin Data
“The Influence of Genetic Factors on Physical Functioning and Exercise in Second Half of Life”
The influence of genetic factors on physical functioning and exercise in second half of life
“Genetic Factors in Physical Activity Levels: A Twin Study”, Maia et al 2002
“Selection, Structure and the Heritability of Behavior”, Stirling et al 2002
“A Genome-Wide Scan of 1842 DNA Markers for Allelic Associations With General Cognitive Ability: A Five-Stage Design Using DNA Pooling and Extreme Selected Groups”, Plomin et al 2001
“Individual Differences in Response to Regular Physical Activity”, Bouchard & Rankinen 2001
Individual differences in response to regular physical activity
“Heritability Estimates versus Large Environmental Effects: The IQ Paradox Resolved”, Dickens & Flynn 2001
Heritability estimates versus large environmental effects: The IQ paradox resolved
“Nonshared Environment: A Theoretical, Methodological, and Quantitative Review”, Turkheimer & Waldron 2000
Nonshared environment: A theoretical, methodological, and quantitative review
“Three Laws of Behavior Genetics and What They Mean”, Turkheimer 2000
“Early Observations of Genetic Diseases”, Urban 1999
“Comparing the Biological and Cultural Inheritance of Personality and Social Att”, Eaves et al 1999
Comparing the biological and cultural inheritance of personality and social att
“Causality in Complex Systems”, Wagner 1999
“Chapter 25: Liability-Threshold Model”, Lynch & Walsh 1998
“Poverty and Behavior: Are Environmental Measures Nature and Nurture?”, Rowe & Rodgers 1997
Poverty and Behavior: Are Environmental Measures Nature and Nurture?
“Francis Galton: His Approach to Polygenic Disease”, Galton & Galton 1997
“An Examination of Genotype-Environment Interactions for Academic Achievement in an US National Longitudinal Survey”, Ord & Rowe 1997
“A ‘Second Front’ in Soviet Genetics: The International Dimension of the Lysenko Controversy, 1944–1947”, Krementsov 1996
“Population Stratifications Can Cause False Positive Linkage Results If Founders Are Untyped”
Population stratifications can cause false positive linkage results if founders are untyped
“Barbara Stoddard Burks: Pioneer Behavioral Geneticist and Humanitarian [Ch15]”, King et al 1996
Barbara Stoddard Burks: Pioneer Behavioral Geneticist and Humanitarian [ch15]
“Galton Lecture: Behavior Genetic Studies of Intelligence, Yesterday and Today: the Long Journey from Plausibility to Proof”, Bouchard 1996
“Arguments About Aborigines: Australia and the Evolution of Social Anthropology: Chapter 7, Conceptions and Misconceptions”, Hiatt 1996
“Conceptions and Misconceptions § Difficulties in Understanding Sex”, Hiatt 1996 (page 21)
Conceptions and Misconceptions § Difficulties in understanding sex
“Separate Social Worlds of Siblings”, Hetherington et al 1994
“Smoking and Sports Participation”, Koopmans et al 1994
“Nature-Nurture Reconceptualized in Developmental Perspective: A Bioecological Model”, Bronfenbrenner & Ceci 1994
Nature-nurture reconceptualized in developmental perspective: A bioecological model
“The Lifetime History of Major Depression in Women: Reliability of Diagnosis and Heritability”, Kendler et al 1993
The Lifetime History of Major Depression in Women: Reliability of Diagnosis and Heritability
“Evolution of the Nature~nurture Issue in the History of Psychology”, PsycBooks 1993
Evolution of the nature~nurture issue in the history of psychology
“Heritability of Interests: a Twin Study”, Lykken et al 1993
“Dyslexia Subtypes, Genetics, Behavior, and Brain Imaging”, Lubs et al 1991
“The Response to Long-Term Overfeeding in Identical Twins”, Bouchard et al 1990b
“The Importance of Twin Studies for Individual Differences Research”
The Importance of Twin Studies for Individual Differences Research
“Confirming Unexpressed Genotypes for Schizophrenia: Risks in the Offspring of Fischer’s Danish Identical and Fraternal Discordant Twins”, Gottesman & Bertelsen 1989
“Genetic and Environmental Determinants of Musical Ability in Twins”
Genetic and environmental determinants of musical ability in twins
“Genes, Culture and Personality: An Empirical Approach”, Eaves et al 1989
“Behavior Genetics Association Abstract”
“Resemblances of Parents and Twins in Sports Participation and Heart Rate”, Boomsma et al 1989
Resemblances of parents and twins in sports participation and heart rate
“Behavior Genetics Association Abstracts”
“Offspring of Twin Pairs Discordant for Psychiatric Illness”, Bertelsen & Gottesman 1986
“A Twin Study of Human Obesity”, Stunkard et al 1986b
“Storm Over Biology: Essays on Science, Sentiment, and Public Policy”, Davis et al 1986
Storm Over Biology: Essays on Science, Sentiment, and Public Policy
“Multiple Regression Analysis of Twin Data Obtained from Selected Samples”
Multiple regression analysis of twin data obtained from selected samples
“The Study of Temperament: Changes, Continuities and Challenges”, Plomin & Dunn 1986
The Study of Temperament: Changes, Continuities and Challenges
“Geneticists and Race”, Provine 1986
“Temperament (PLE: Emotion): Early Developing Personality Traits”, Buss et al 1984
Temperament (PLE: Emotion): Early Developing Personality Traits
“The Colorado Adoption Project”
“Identical Twins Reared Apart: Reanalysis or Pseudo-Analysis?”, Bouchard 1982
Identical Twins Reared Apart: Reanalysis or Pseudo-analysis?
“Genetic and Environmental Influences on the Development of Piagetian Logico-Mathematical Concepts and Other Specific Cognitive Abilities: A Twin Study”, Garfinkle 1982
“Cigarette Smoking, Use of Alcohol, and Leisure-Time Physical Activity among Same-Sexed Adult Male Twins”, Kaprio et al 1981
“Twins: Black and White”, Osborne 1980
“Personality and Heredity: An Introduction to Psychogenetics”, Wells 1980
“A Note on the Heritability of Memory Span”
“Twin Method: Defense of a Critical Assumption”, Scarr & Carter-Saltzman 1979
“Infantile Autism: A Genetic Study Of 21 Twin Pairs”, Folstein & Rutter 1977
“Social Genetics”
“Heritability of IQ [Letter in Response to ‘The Heritability Hang-Up’, by M. Feldman and R. C. Lewontin]”, Plomin & DeFries 1976
“The Multifactorial/threshold Concept-Uses and Misuses”
“Issues in the Genetics of Social Behavior”
“Sibling Deidentification”, Schachter et al 1976
“The Determinants of Earnings: Genetics, Family, and Other Environments: A Study of White Male Twins”
The Determinants of Earnings: Genetics, Family, and Other Environments: A Study of White Male Twins
“The Professor, The Institute and DNA”, Dubos 1976
Heredity, Environment, & Personality: A Study of 850 Sets of Twins, Loehlin & Nichols 1976
Heredity, Environment, & Personality: A Study of 850 Sets of Twins
“The Meaning of Heritability in the Behavioral Sciences”, Jensen 1975b
“A Test of a Primary Bias in Twin Studies with respect to Measured Ability”
A test of a primary bias in twin studies with respect to measured ability
“Biological Aspects of a High Socio-Economic Group II. IQ Components and Social Mobility”, Gibson & Mascie-Taylor 1973b
Biological aspects of a high socio-economic group II. IQ Components and Social Mobility
“The Multifactorial Model for the Inheritance of Liability to Disease and Its Implications for Relatives at Risk”
Genetics, Environment, and Behavior: Implications for Educational Policy, Ehrman et al 1972
Genetics, Environment, and Behavior: Implications for Educational Policy
“Behavior and Heredity [Open Letter]”, Page 1972c
“Heritability of Liability and Concordance in Monozygous Twins”, Smith 1970
Heritability of liability and concordance in monozygous twins
“Schizophrenia: Evidence for the Major Gene Hypothesis”, Elston & Campbell 1970
“Behavioral Genetics [Annual Review]”, McClearn 1970
“Biology and Behavior: Genetics”, Glass 1968
“The Effect of Assortative Mating on the Genetic Composition of a Population”, Crow & Felsenstein 1968
The effect of assortative mating on the genetic composition of a population
“The Inheritance of Liability to Diseases With Variable Age of Onset, With Particular Reference to Diabetes Mellitus”, Falconer 1967
“Hereditary Influences on Vocational Preferences As Shown by Scores of Twins on the Minnesota Vocational Interest Inventory”, Vandenberg & Stafford 1967
“Possible Transfer of Metallurgical and Astronomical Approaches to Problem of Environment versus Ethnic Heredity”, Shockley 1966
“Commentary on R. A. Fisher‘s Paper on 'The Correlation Between Relatives on the Supposition of Mendelian Inheritance’”, Moran & Smith 1966
“Genetic Factors in Activity Motivation”
“The Inheritance of Liability to Certain Diseases, Estimated from the Incidence among Relatives”
The inheritance of liability to certain diseases, estimated from the incidence among relatives
“Hereditary Components in Vocational Preferences”, Vandenberg & Kelly 1964
“Roots of Behavior: Genetics, Insect, and Socialization in Animal Behavior”, Bliss 1962
Roots of Behavior: Genetics, Insect, and Socialization in Animal Behavior
“Behavior Genetics”, Fuller 1960
“Heredity, Environment, and the Question "How?"”, Anastasi 1958
“The Potential Contribution of Longitudinal Twin Studies: An Appraisal”, Falkner 1957
The potential contribution of longitudinal twin studies: An appraisal
“The Importance of Being Cross-Bred”, Michie & McLaren 1955
Twins: A Study of 3 Pairs of Identical Twins, Burlingham 1952
“Studies of Identical Twins Reared Apart”, Burks & Roe 1949
“The Nature-Nurture Controversy”, Pastore 1949
“The Incidence and Mendelian Transmission of Mid-Digital Hair in Man”, Bernstein & Burks 1942
The incidence and Mendelian transmission of mid-digital hair in man
“Twins: A Study of Heredity and Environment”, Newman et al 1937
“Twin-Similarities in Personality Traits”, Carter 1933
“Statistical Methods in Biology”, Wright 1931
“The Biological Basis of Human Nature”, Jennings 1930
“Chapter 10: The Relative Influence of Nature and Nurture upon Mental Development: A Comparative Study of Foster Parent-Offspring Child Resemblance and True Parent-True Child Resemblance”, Burks 1928b
“Chapter II: Statistical Hazards in Nature-Nurture Investigations”, Burks 1928
Chapter II: Statistical hazards in nature-nurture investigations
“Heredity and Environment”, Jennings 1924
“On the Inheritance of the Mental and Moral Characters in Man, and Its Comparison With the Inheritance of the Physical Characters”, Pearson 1903
“A Mega-Analysis of Genome-Wide Association Studies for Major Depressive Disorder”
A mega-analysis of genome-wide association studies for major depressive disorder
“I’m Not A Look-A-Like!”, Brunelle 2026
“The Lettre Lab: Members”, Lettre 2026
“Behavior Genetic Frameworks of Causal Reasoning for Personality Psychology”
Behavior Genetic Frameworks of Causal Reasoning for Personality Psychology
“Applying Compressed Sensing to Genome-Wide Association Studies”
Applying compressed sensing to genome-wide association studies
“Determination of Nonlinear Genetic Architecture Using Compressed Sensing”
Determination of nonlinear genetic architecture using compressed sensing
“Common Genetic Variants Associated With Cognitive Performance”, Hsu 2026
Common genetic variants associated with cognitive performance
“Big Data: Astronomical or Genomical?”, Stephens et al 2026
“G = E: What GWAS Can Tell Us about the Environment”, Gage et al 2026
“Andrew Wood”
“Genetic Determinants of the Gut Microbiome in UK Twins”
“Gut Microbiome Heritability Is Near-Universal but Environmentally Contingent”
Gut microbiome heritability is near-universal but environmentally contingent
“Jean-Baptiste Pingault”
“The Right Not to Know: When Ignorance Is Bliss but Deadly”
“Serena Sanna”
“Society Is Fixed, Biology Is Mutable”
“Is Bronny James Underrated? Inside the Phenomenon of the NBA Bloodline”
Is Bronny James underrated? Inside the phenomenon of the NBA bloodline
“The Myth of Human Fragility”
“Personality Polygenes, Positive Affect, and Life Satisfaction”
Personality Polygenes, Positive Affect, and Life Satisfaction
“A Newly Discovered Tea Plant Is Caffeine-Free: It Was Found Growing Wild in Fujian Province”
A newly discovered tea plant is caffeine-free: It was found growing wild in Fujian province
“DNA Sequencing Costs: Data”
“The Cost of Sequencing a Human Genome”
“Association of the Anxiogenic and Alerting Effects of Caffeine With ADORA2A and ADORA1 Polymorphisms and Habitual Level of Caffeine Consumption”
“The Texas Twin Project”
“Genome-Wide Association Studies of a Broad Spectrum of Antisocial Behavior”
Genome-Wide Association Studies of a Broad Spectrum of Antisocial Behavior
“Estimating the Heritability of Cognitive Traits across Dog Breeds Reveals Highly Heritable Inhibitory Control and Communication Factors”
“A New Test Can Predict IVF Embryos’ Risk of Having a Low IQ: A New Genetic Test That Enables People Having IVF to Screen out Embryos Likely to Have a Low IQ or High Disease Risk Could Soon Become Available in the US”
“$400 Million Investment Programme Positions Ireland for Global Leadership in Genomic Research and Advanced Life Sciences”
“Additive Genetic Variation in Schizophrenia Risk Is Shared by Populations of African and European Descent”
“The Infinitesimal Model: Definition, Derivation, and Implications”
The infinitesimal model: Definition, derivation, and implications
“Xist Ribonucleoproteins Promote Female Sex-Biased Autoimmunity”
Xist ribonucleoproteins promote female sex-biased autoimmunity
“Intelligence Indexes Generalist Genes for Cognitive Abilities”
Intelligence indexes generalist genes for cognitive abilities
“Is There a Genetic Link to Being an Extremely Good Boy?”
“Now You Can Sequence Your Whole Genome for Just $200”
“The Importance of Heritability in Psychological Research: The Case of Attitudes”
The Importance of Heritability in Psychological Research: The Case of Attitudes
doctorveera
Sort By Magic
Annotations sorted by machine learning into inferred 'tags'. This provides an alternative way to browse: instead of by date order, one can browse in topic order. The 'sorted' list has been automatically clustered into multiple sections & auto-labeled for easier browsing.
Beginning with the newest annotation, it uses the embedding of each annotation to attempt to create a list of nearest-neighbor annotations, creating a progression of topics. For more details, see the link.
collectibles
education-metrics
soviet-genetics genetic-disease historical-genetics lysenko-controversy medical-genetics genetics-history
understanding-bias
dna-decay
disease-susceptibility
population-genetics
facial-aging
dna-profiling
polygenic-risk
recessive-allele
genetic-admixture
phenotypic-plasticity
education-outcomes
Wikipedia (8)
Miscellaneous
/doc/genetics/heritable/2023-abdellaoui-figure1-gwassamplesizeandhitcount2007to2022.jpg/doc/genetics/heritable/2023-wesseldijk-figure1-musicheritabilities.jpg/doc/genetics/heritable/2022-patel-figure2-heartdiseasepredictionbypolygenicscores.png/doc/genetics/heritable/2021-hart-figure2-geneticconfoundingdag.jpg/doc/genetics/heritable/2021-owen-figure1-rapidwgsmedicaltimeline.png/doc/genetics/heritable/2020-08-nhgri-sequencingcostpergenome.jpg/doc/genetics/heritable/2020-01-24-meme-mickeymouse-perishlikeadog-genetics.jpg/doc/genetics/heritable/2020-york-figure3-mediaconsumptionheritability.png/doc/genetics/heritable/2019-border-figure2-failuretoreplicatedepressioncandidategenehits.jpg/doc/genetics/heritable/2019-border-figure3-failuretoreplicategenelevelcandidategenehits.jpg/doc/genetics/heritable/2019-maclean-figure1-withincrossbreeddogheritabilities.jpg/doc/genetics/heritable/2018-khan-figure1-consumergenomesequencing20132018.jpg/doc/genetics/heritable/2018-wray-figure1-betweenindividualpolygenicdifferences.jpg/doc/genetics/heritable/2016-ge-ukbiobank-snpheritability.csv/doc/genetics/heritable/2016-hill-ses-health-geneticcorrelations.jpg/doc/genetics/heritable/2015-dochtermann-figure2-heritabilityofanimalpersonality.jpg/doc/genetics/heritable/2015-germine-figure2-acemodeloffacialattractivenessratings.jpg/doc/genetics/heritable/2015-polderman-matchctglabnl-recreationleisure-modelwordcloud.png/doc/genetics/heritable/2010-hatemi-table3-politicalattitudesheritability.png/doc/genetics/heritable/2001-provine-originstheoreticalpopulationgenetics.pdf/doc/genetics/heritable/1998-lynchwalsh-geneticsquantitativetraits-ch27-reml.pdf/doc/genetics/heritable/1995-lubinski-assessingindividualdifferenceshumanbehavior.pdf#page=250/doc/genetics/heritable/1994-plomin-geneticsandexperience.pdf/doc/genetics/heritable/1993-tesser-table1-politicalattitudesheritability.png/doc/genetics/heritable/1986-fuller-perspectivesinbehaviorgenetics.pdf/doc/genetics/heritable/1986-malina-sportandhumangenetics.pdf/doc/genetics/heritable/1983-fuller-behaviorgeneticsprinciplesandapplications.pdf/doc/genetics/heritable/1982-cattell-inheritanceofpersonalityability.pdf/doc/genetics/heritable/1980-juelnielsen-individualandenvironmentmza.pdf/doc/genetics/heritable/1980-osborne-twinsblackandwhite-appendix.ods/doc/genetics/heritable/1979-nichols-heredityandenvironment.pdf/doc/genetics/heritable/1977-halsey-heredityandenvironment.pdf/doc/genetics/heritable/1973-dobzhansky-geneticdiversityandhumanequality.pdf/doc/genetics/heritable/1973-mcclearn-introductiontobehavioralgenetics.pdf/doc/iq/1973-gibson-table2-correlationbetweeniqandsocioeconomistatusbetweenfathersoninengland.jpg/doc/genetics/heritable/1962-shields-monozygotictwinsbroughtupapart.pdf/doc/genetics/heritable/gwern-iq-selection-benyamin2014-polygenicscore-betas.png/doc/genetics/heritable/gwern-iq-gwas-pgs-measurementerror.jpghttp://biology-web.nmsu.edu/~houde/genetics%20of%20aggression.pdfhttps://ecommons.cornell.edu/items/52ad998f-6ad3-485f-93ea-899a0a40fe64https://inquisitivebird.substack.com/p/where-parents-make-a-differencehttps://journals.sagepub.com/doi/full/10.1177/09636625241245030https://knowablemagazine.org/article/living-world/2021/the-curious-case-shrinking-genomehttps://onlinelibrary.wiley.com/doi/pdf/10.1002/ajmg.b.32333https://onlinelibrary.wiley.com/doi/pdf/10.1002/ajmg.b.32363https://onlinelibrary.wiley.com/doi/pdf/10.1002/ajmg.b.32388https://onlinelibrary.wiley.com/doi/pdf/10.1002/ajmg.b.32419https://pdfs.semanticscholar.org/2cb9/e5fe8009b934faa127778bf6aae028e30b62.pdfhttps://pdfs.semanticscholar.org/b15f/026aadf93744168fe9992cb86002a85647d2.pdfhttps://royalsocietypublishing.org/doi/10.1098/rstb.2008.0037https://www.businesswire.com/news/home/20181129005208/en/Ancestry-Breaks-November-Sales-Recordhttps://www.frontiersin.org/articles/10.3389/fcvm.2019.00175/fullhttps://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2021.722187/fullhttps://www.lesswrong.com/posts/cit3HYXehBsr4d36Q/open-thread-january-15-31-2012#rWkZ3TGRsL7DhSsNRhttps://www.theringer.com/nba/2024/4/3/24119579/twinning-time-brook-robin-lopez-amen-ausar-thompsonhttps://www.theturnertwins.co.uk/fitness/40-vs-20-minute-workout
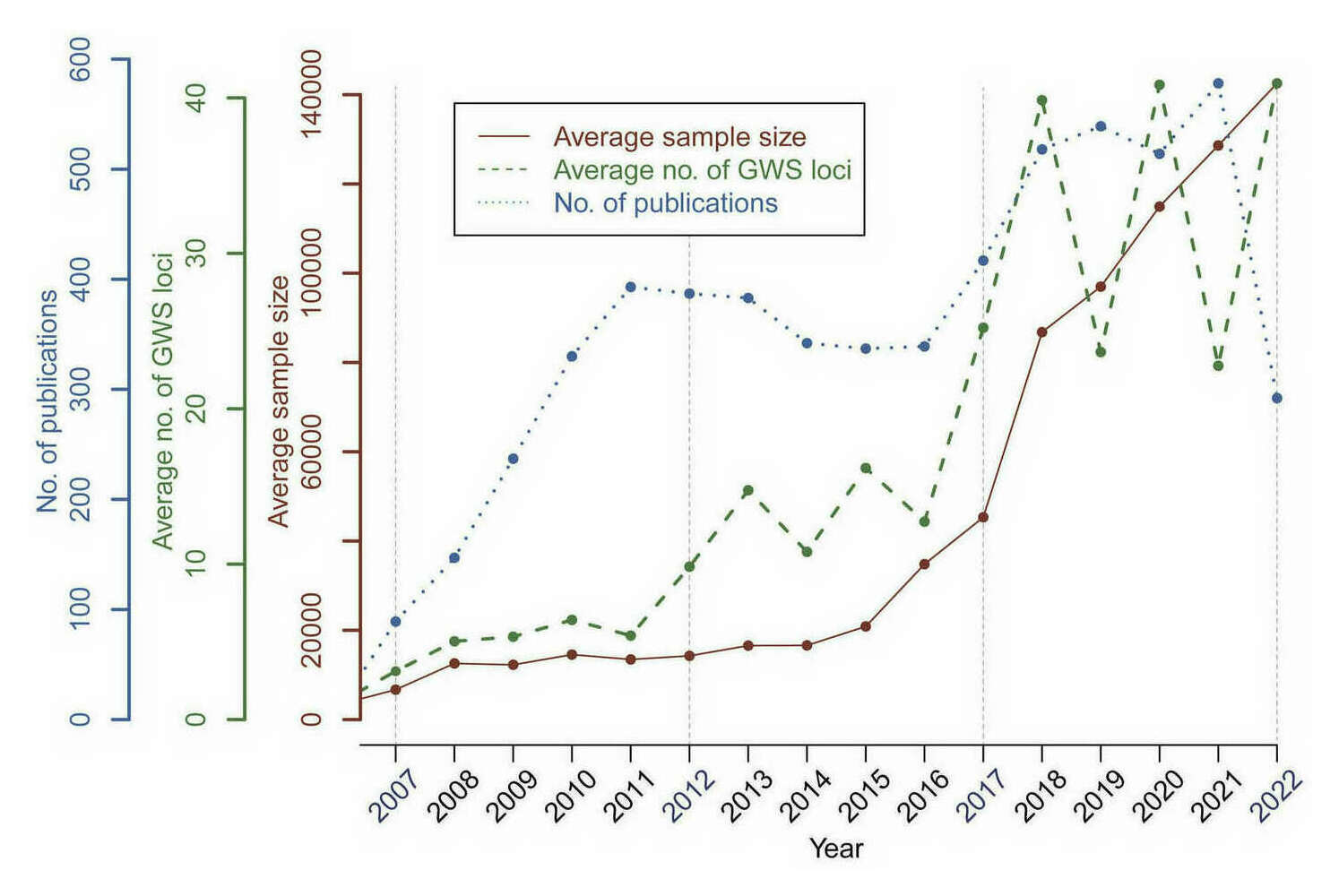{kind=link}
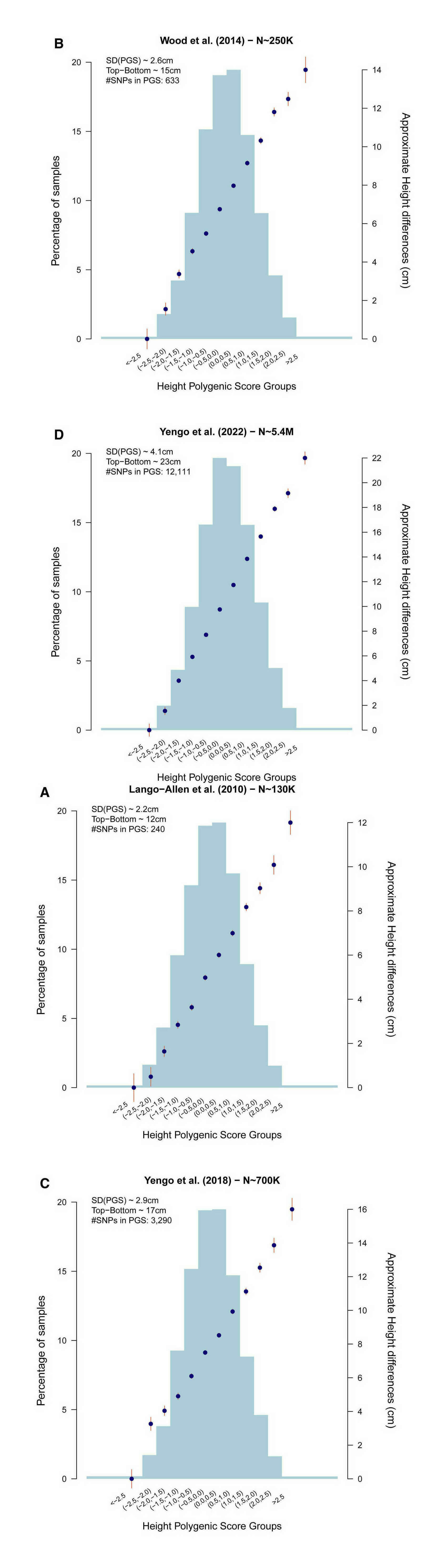{kind=link}
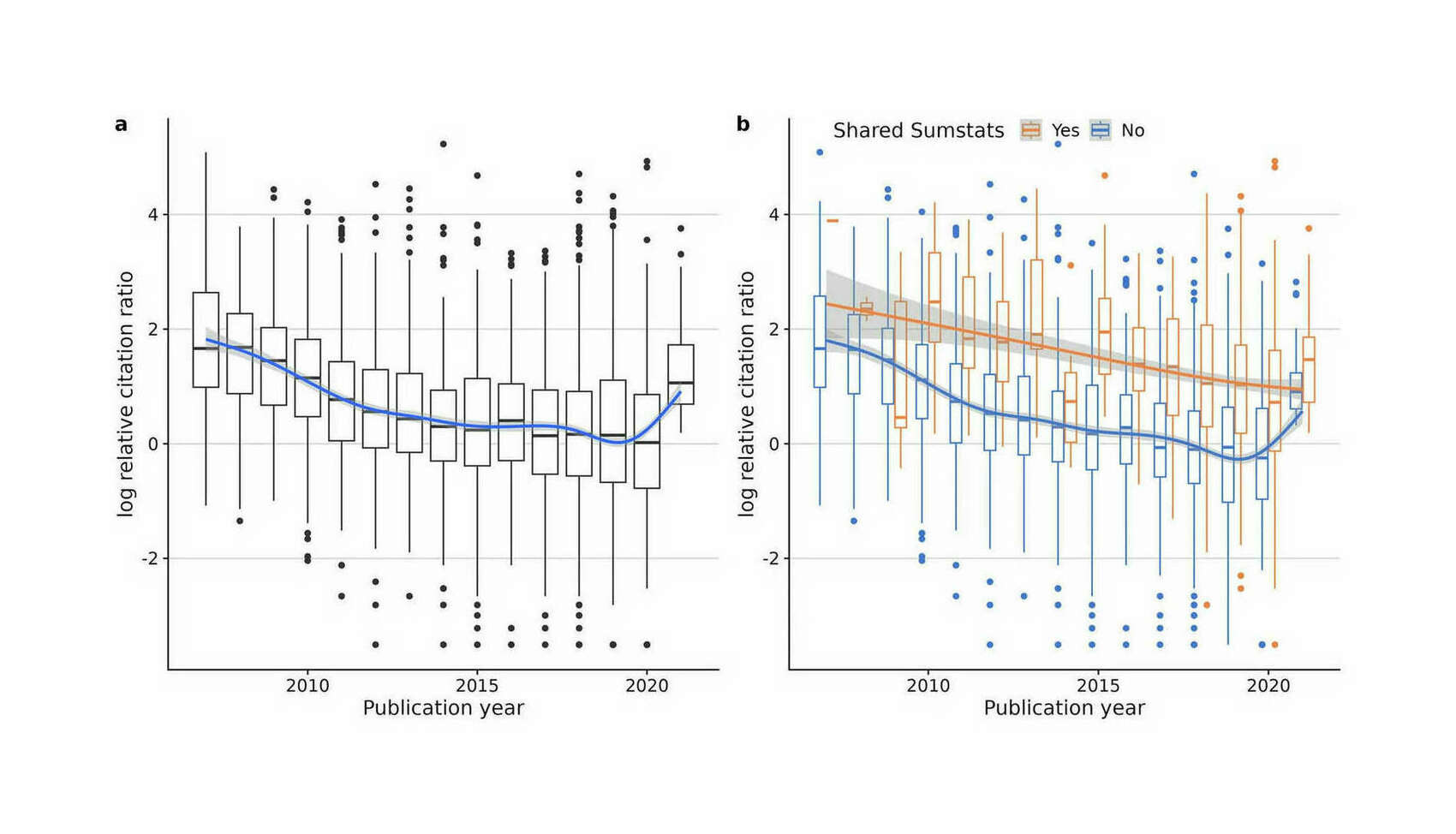{kind=link}
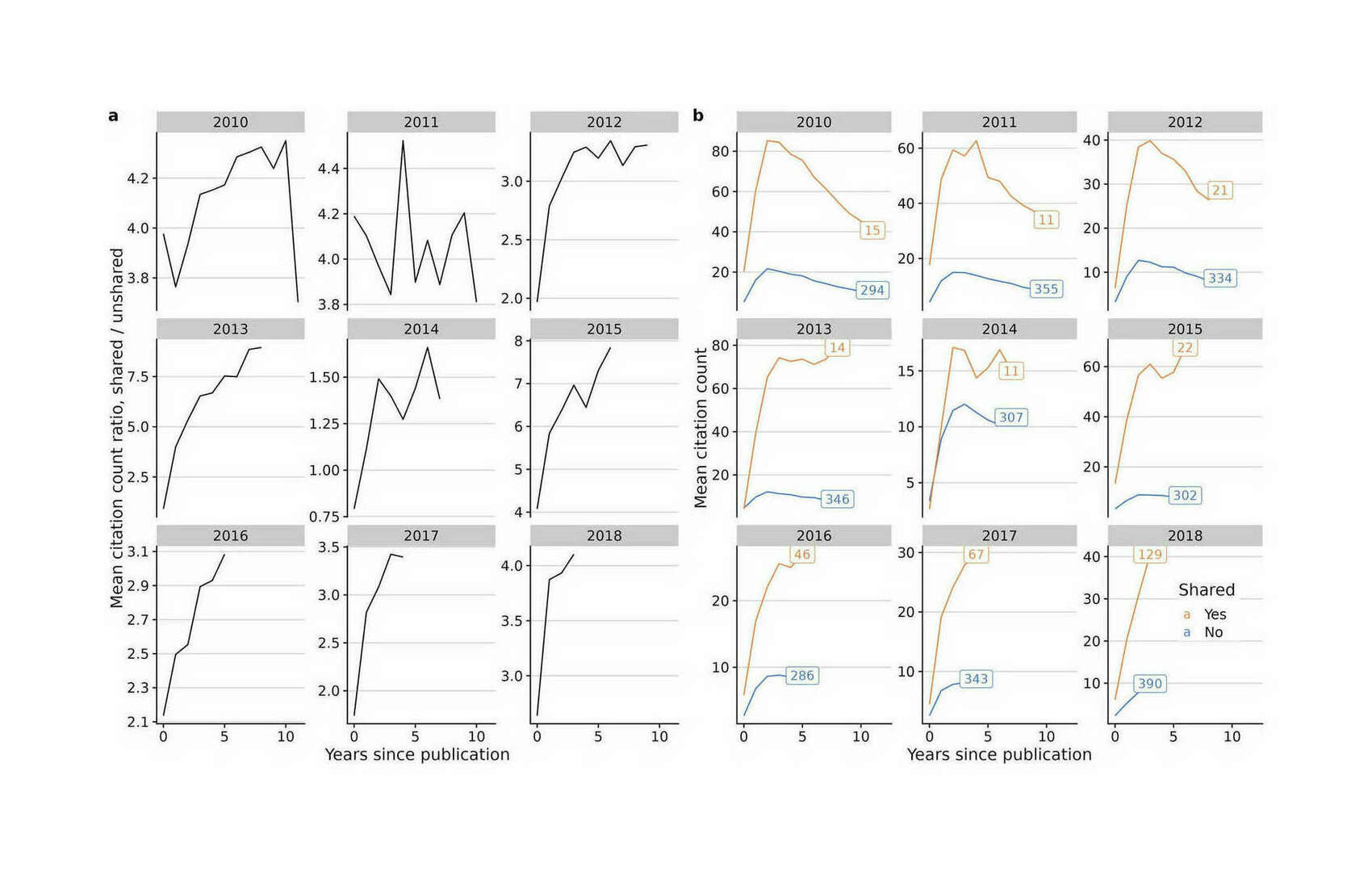{kind=link}
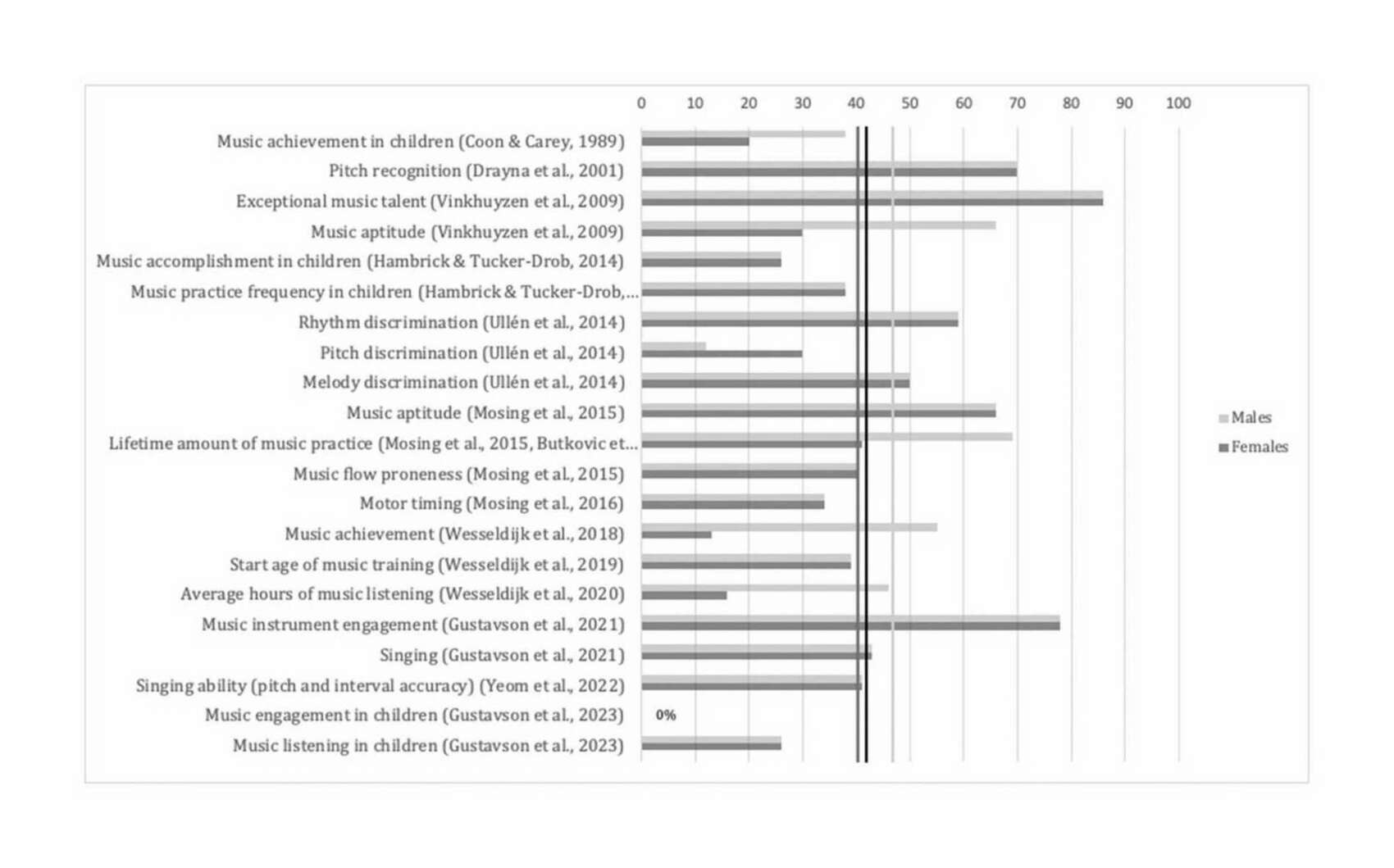{kind=link}
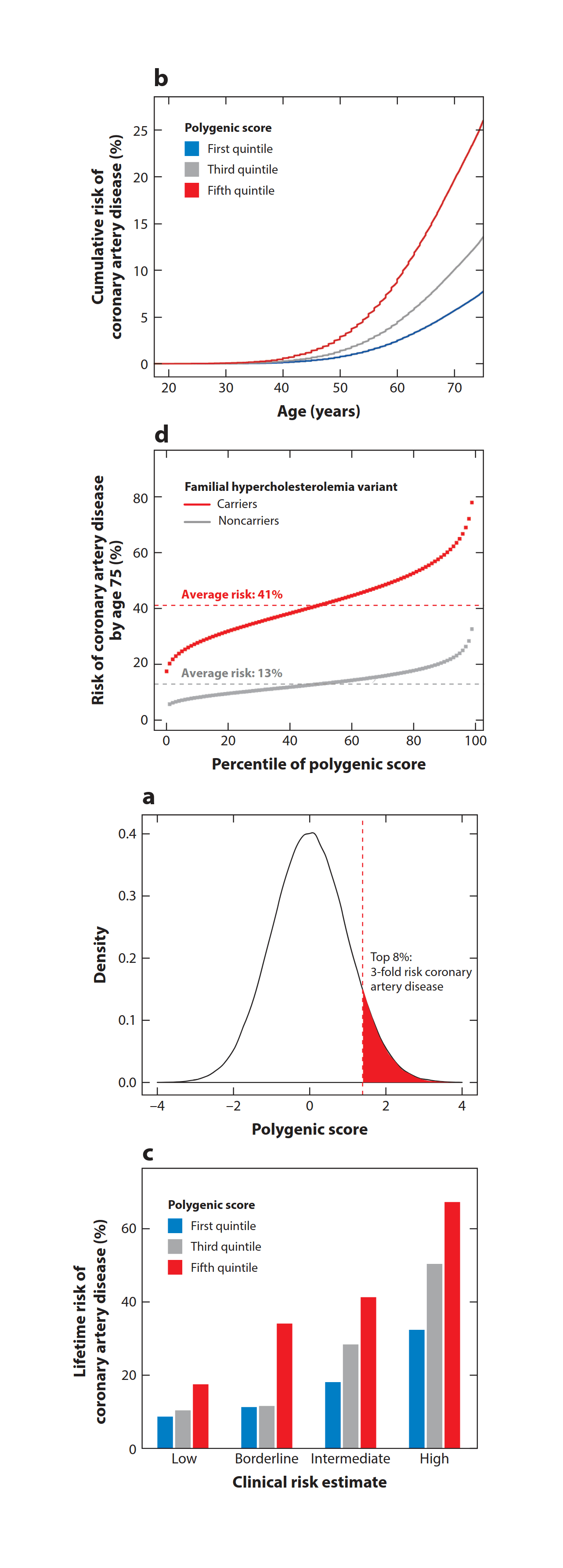{kind=link}
{kind=link}
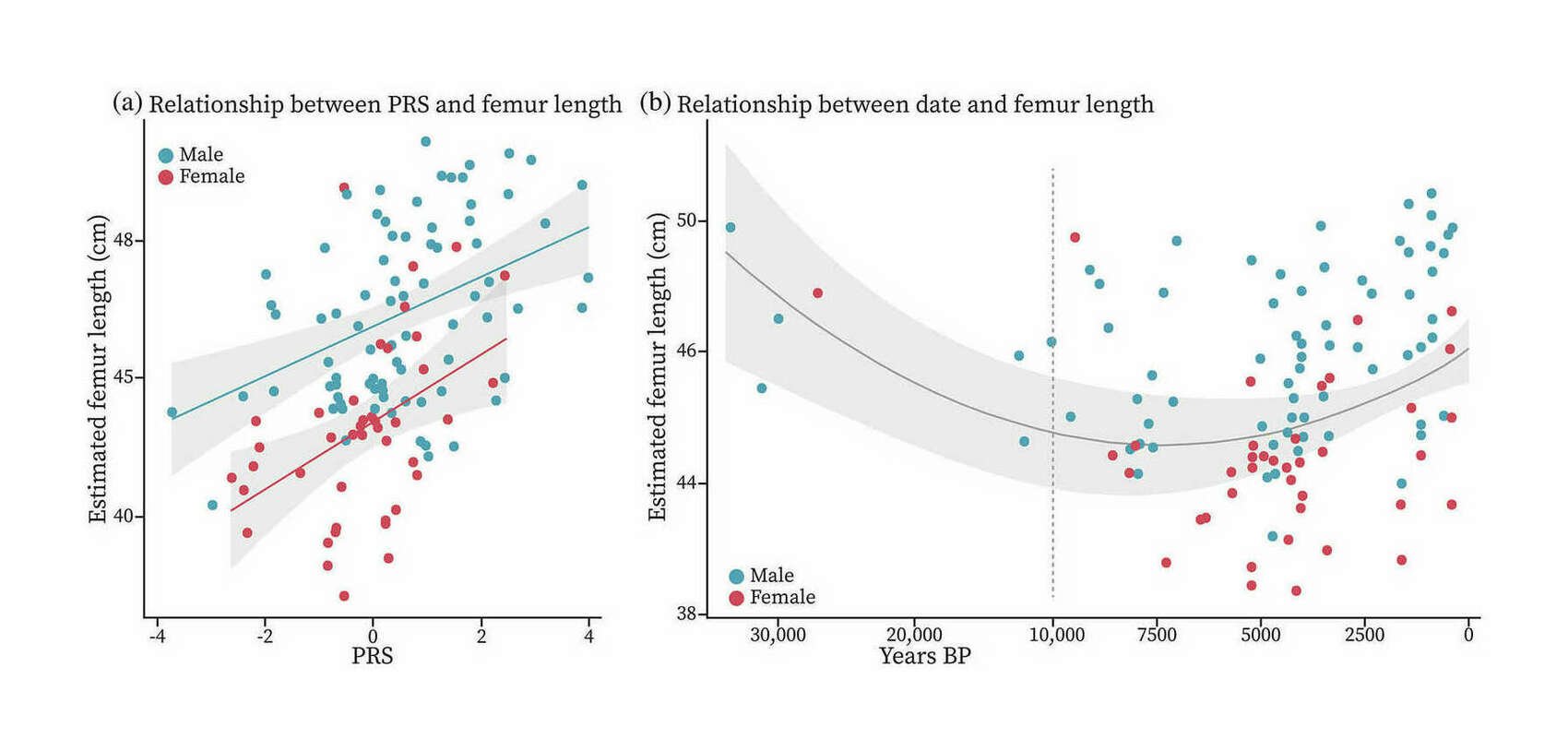{kind=link}
{kind=link}
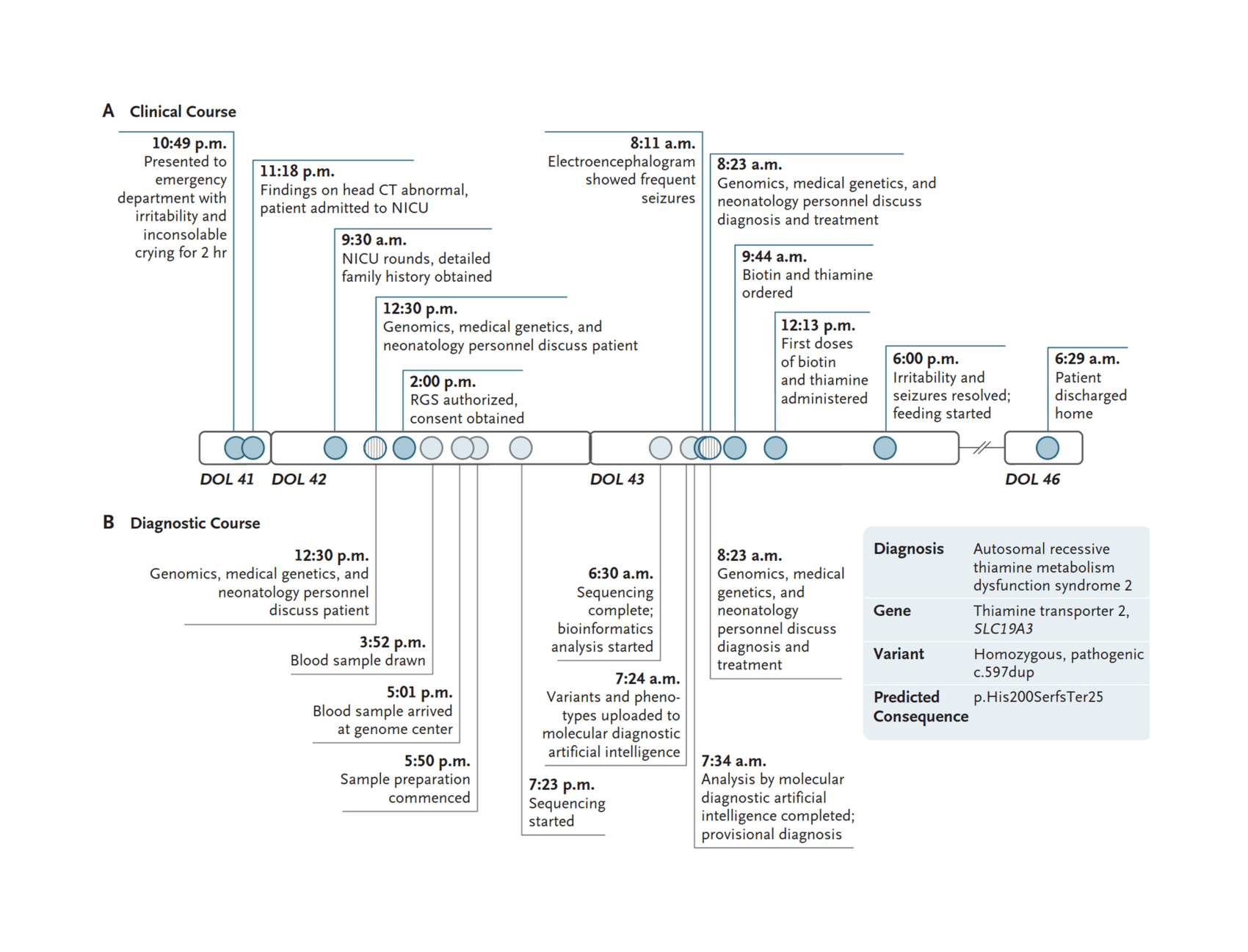{kind=link}
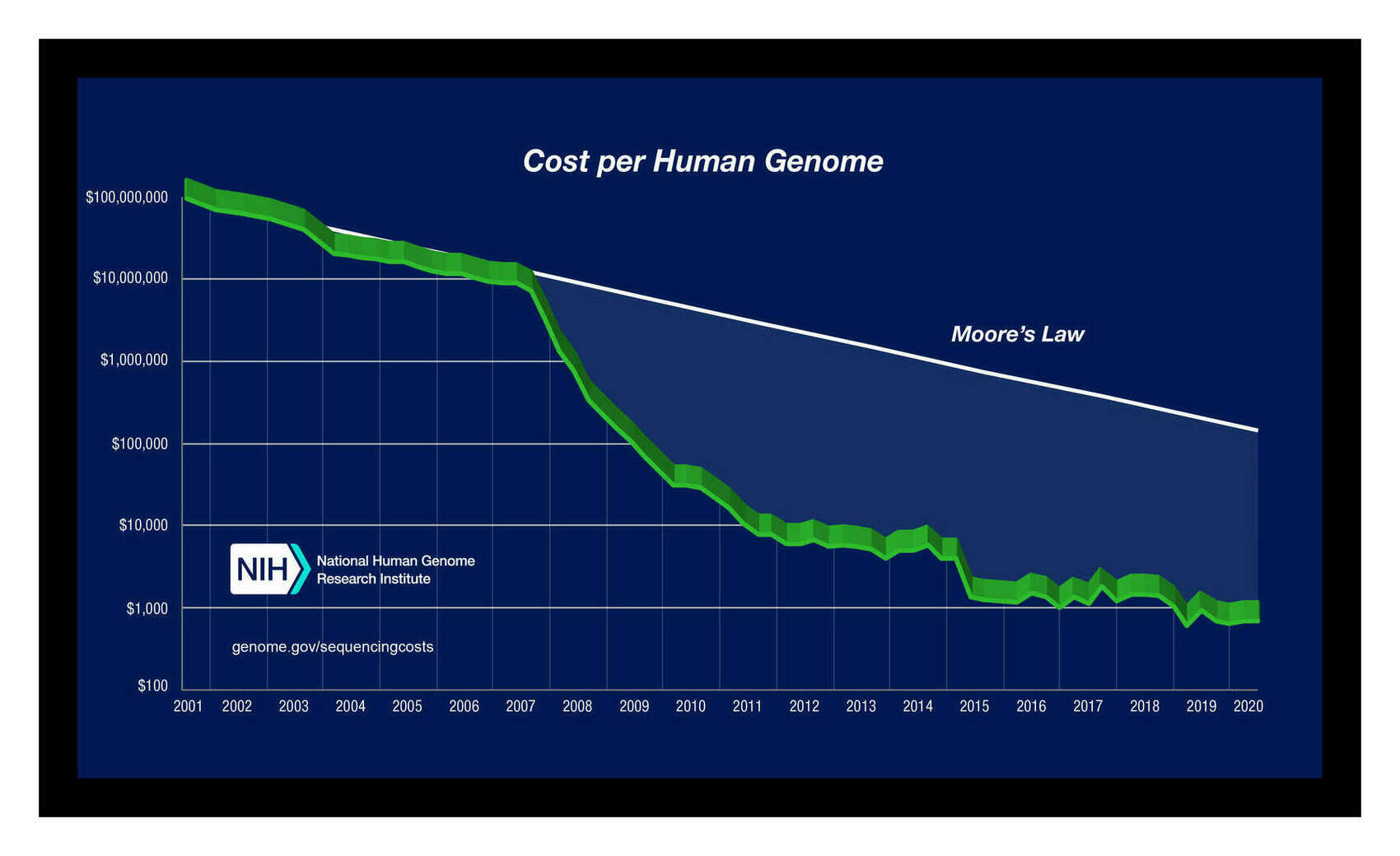{kind=link}
{kind=link}
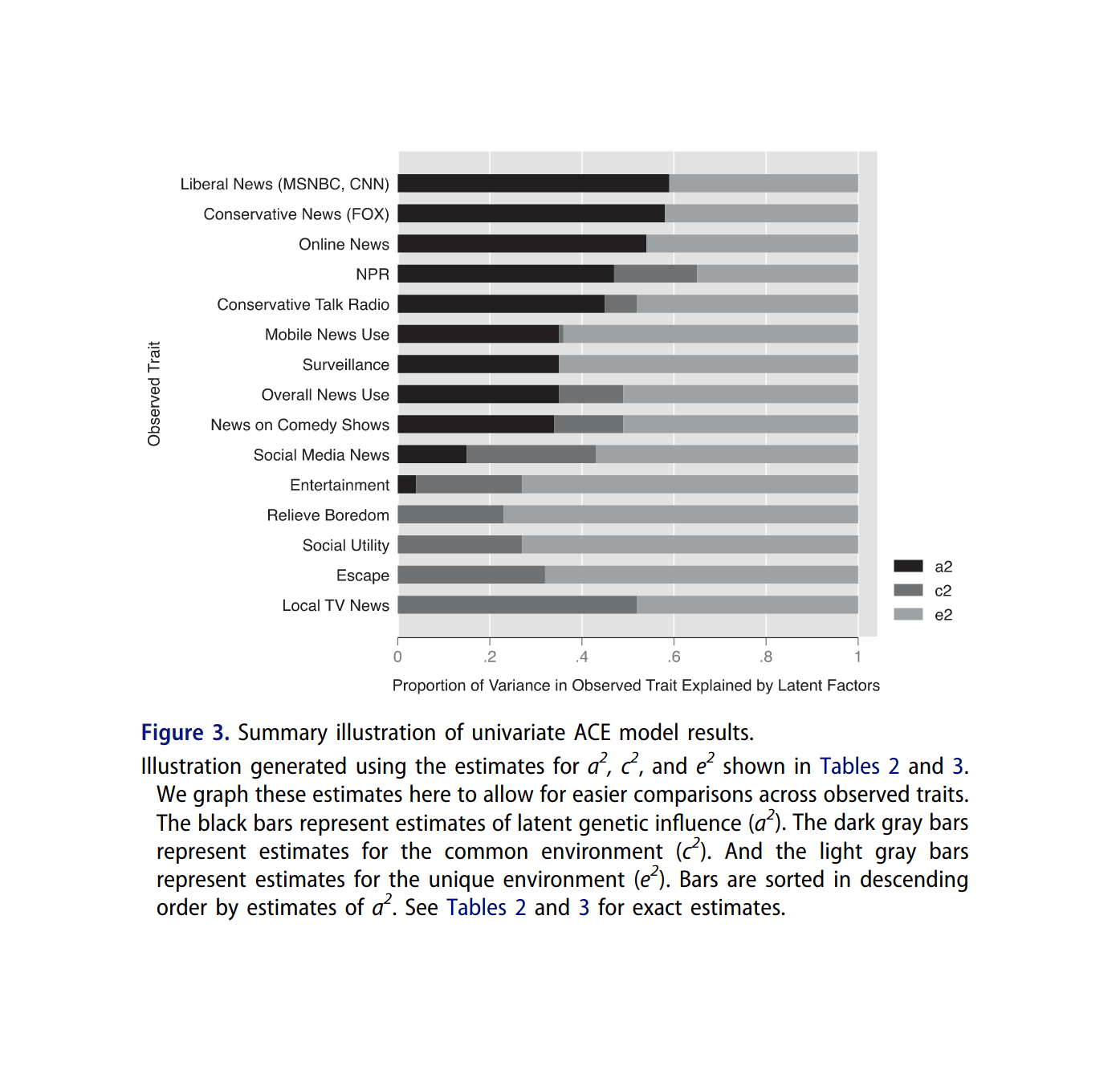{kind=link}
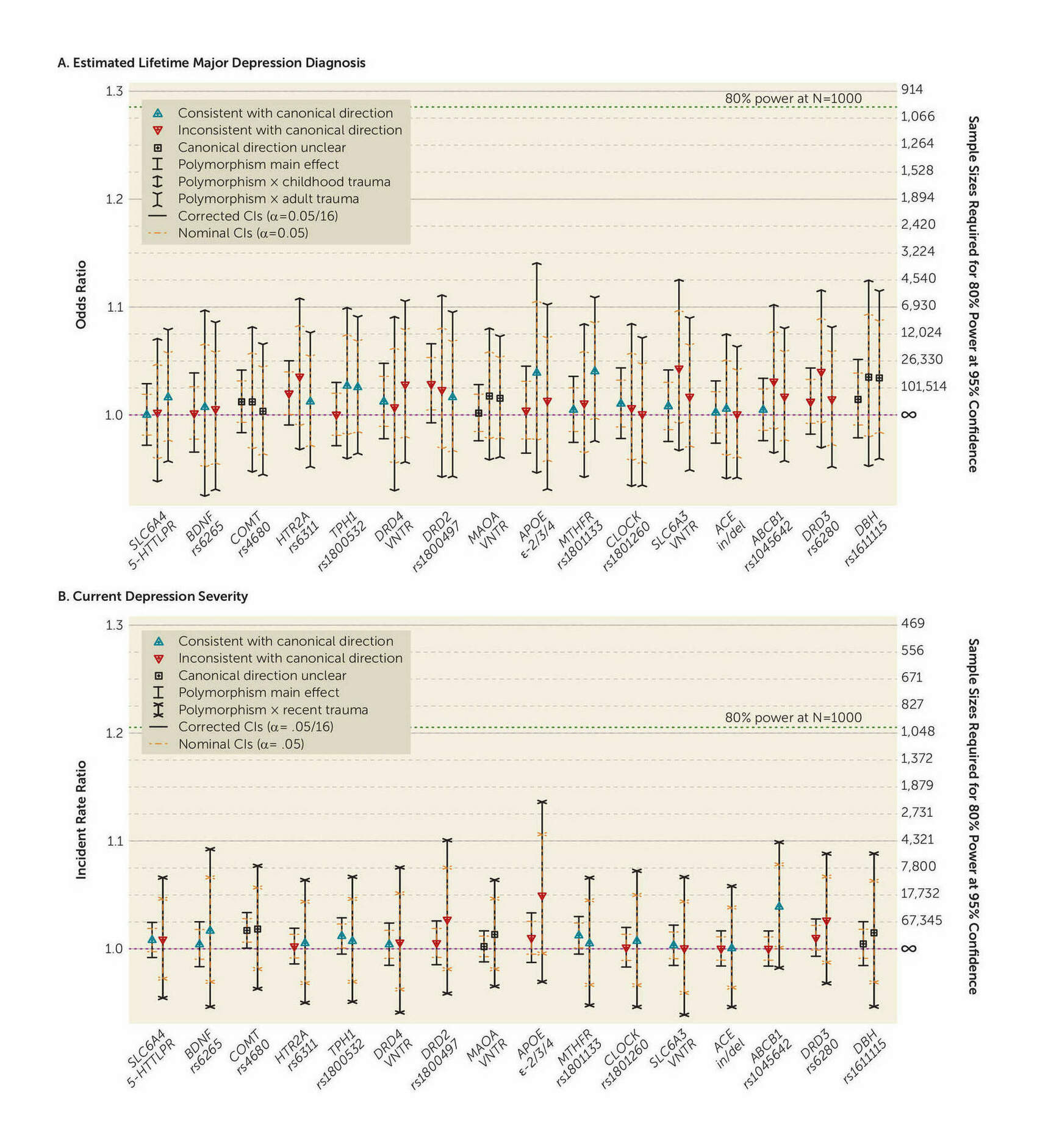{kind=link}
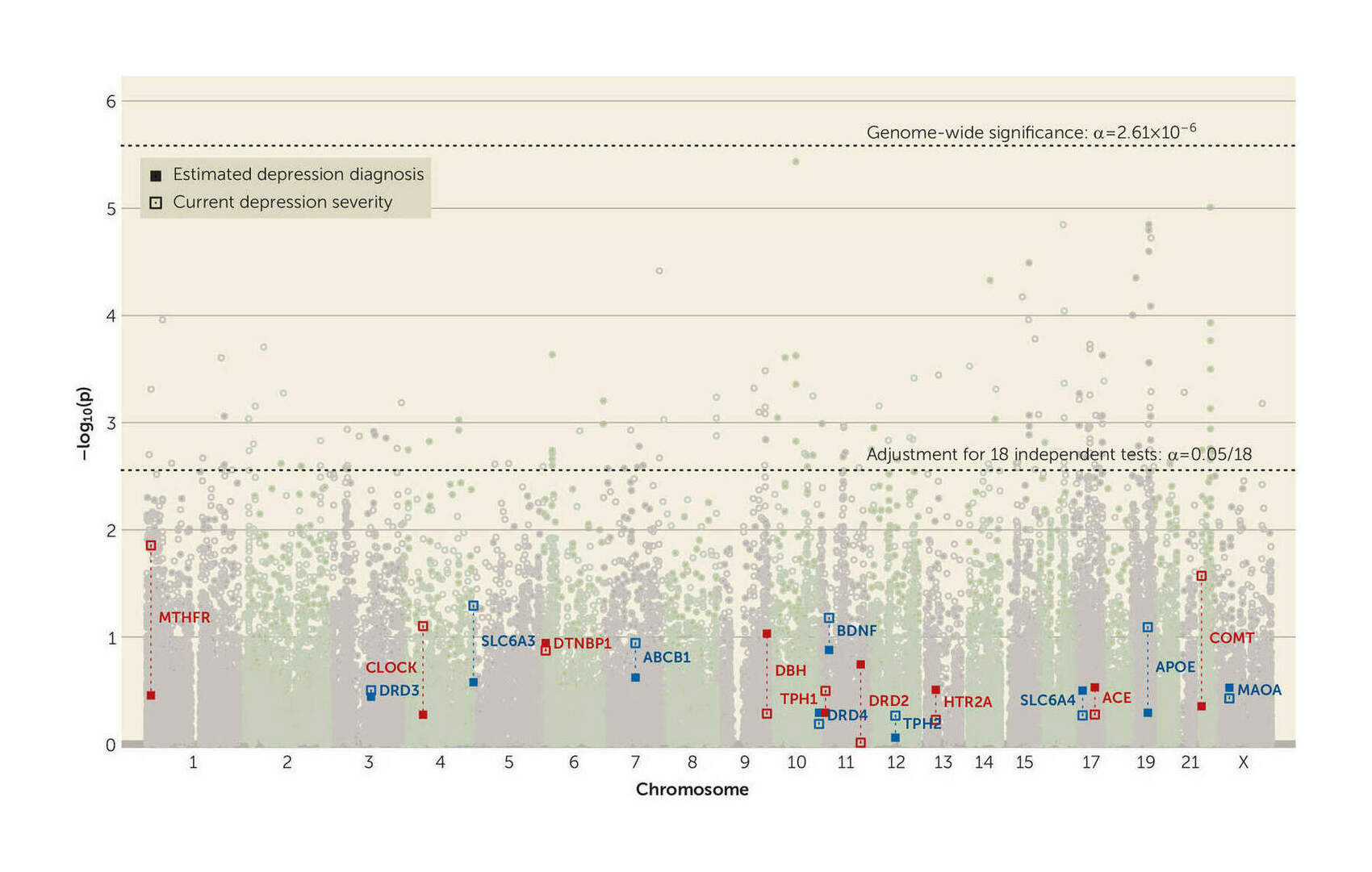{kind=link}
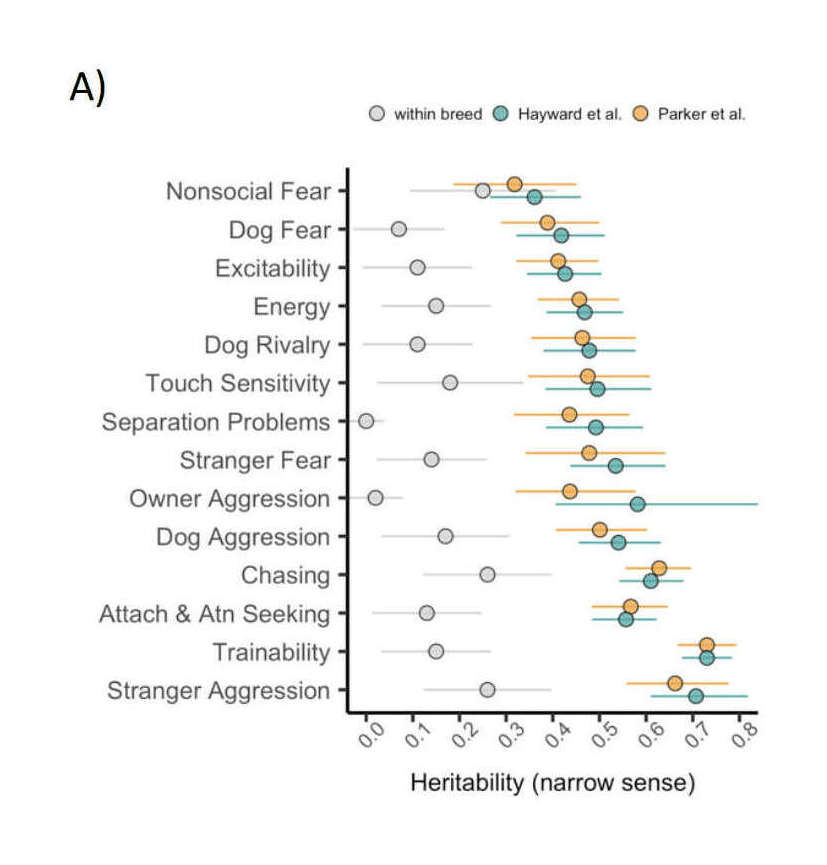{kind=link}
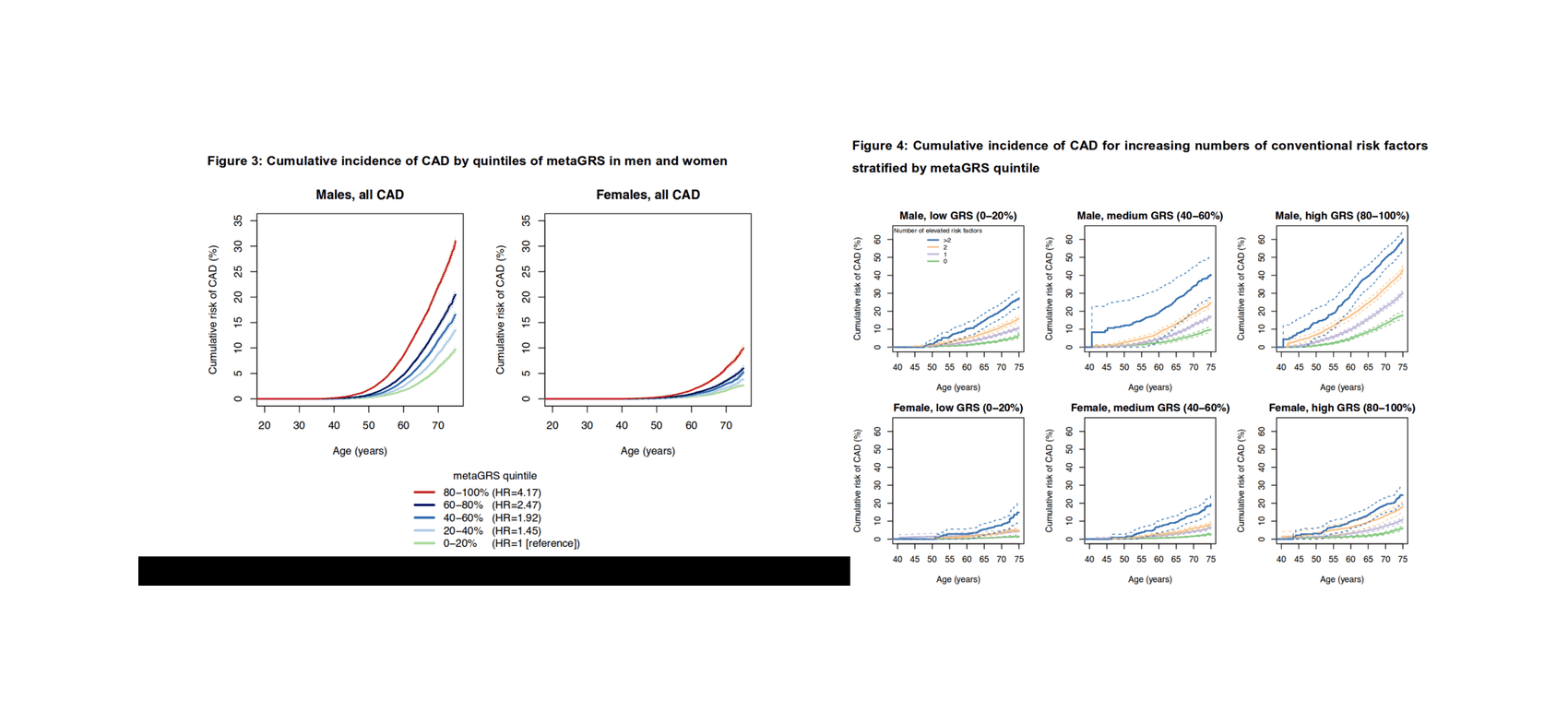{kind=link}
{kind=link}
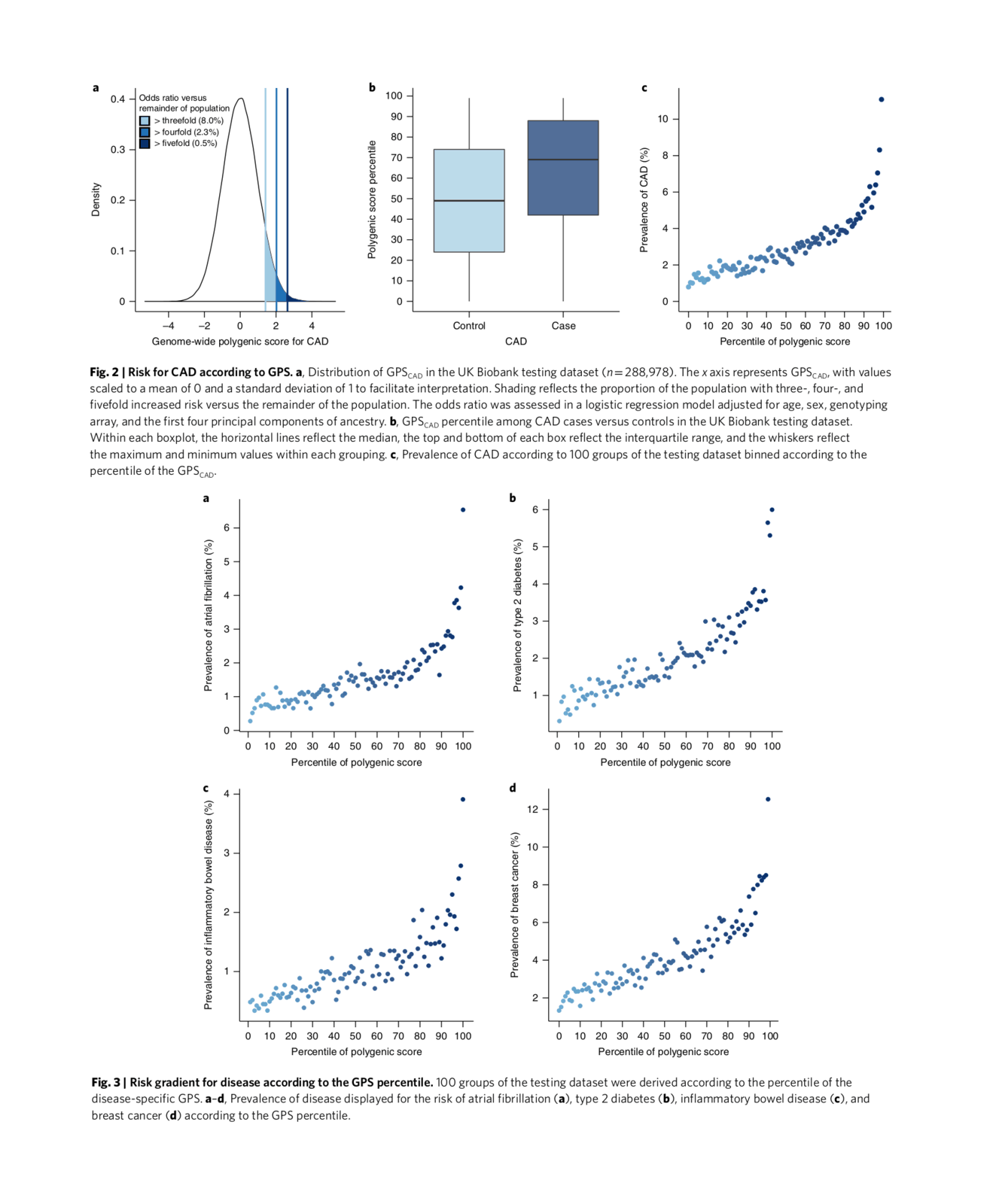{kind=link}
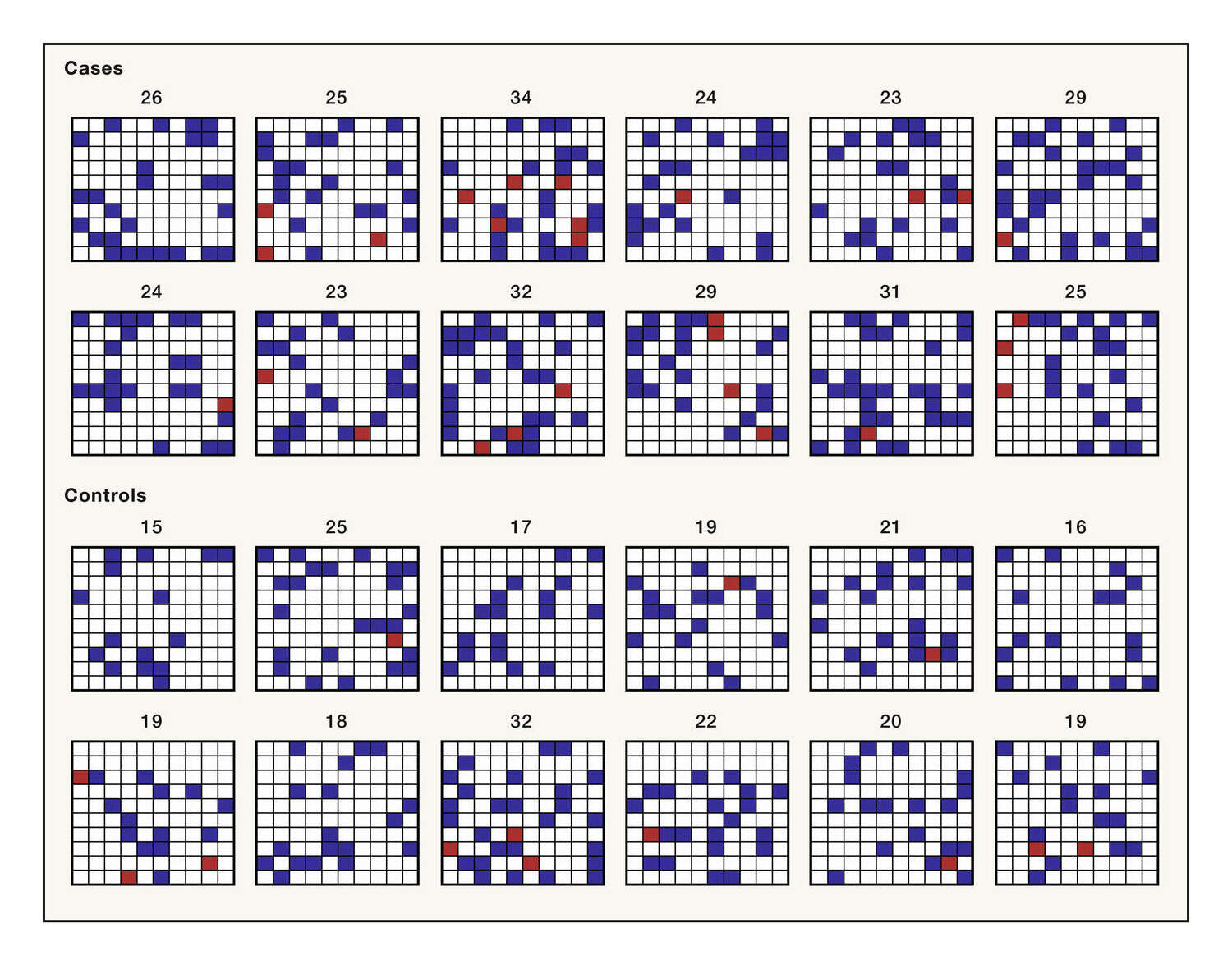{kind=link}
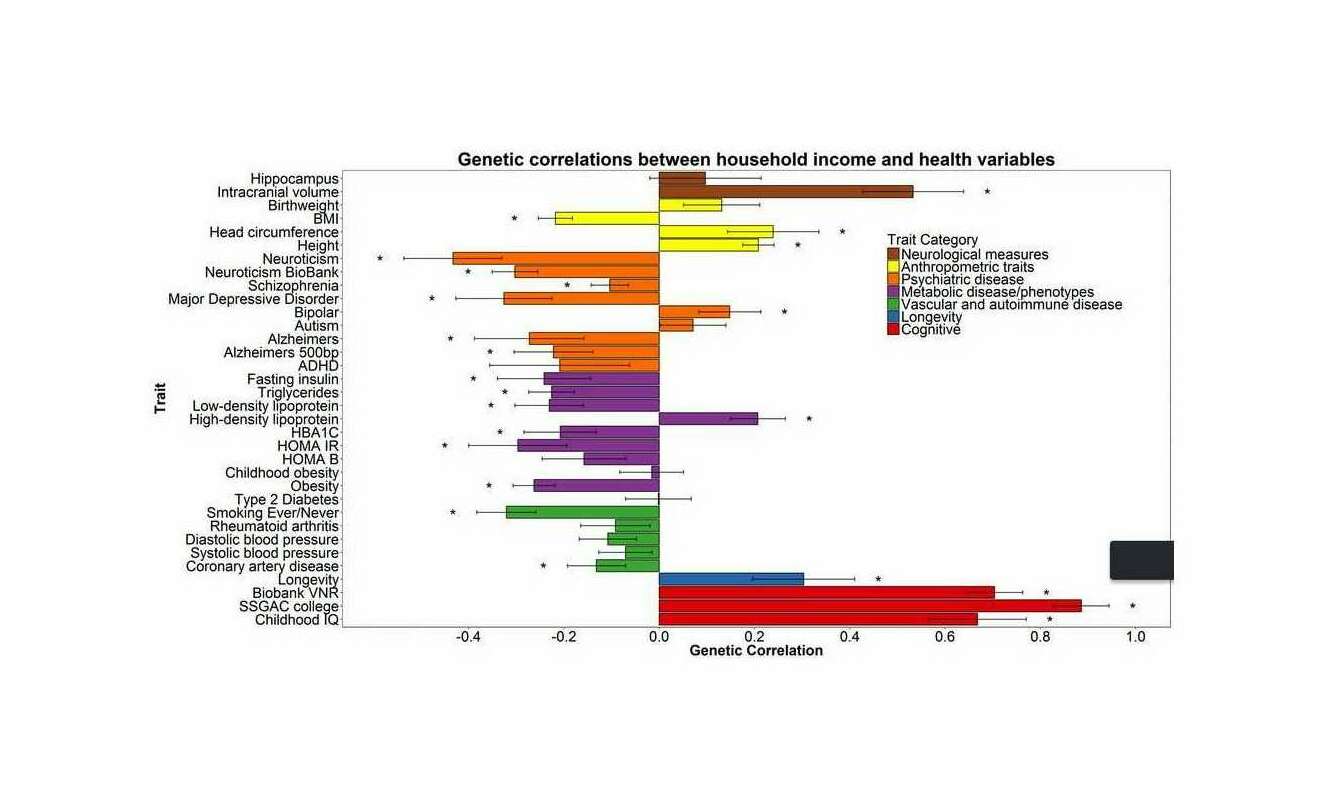{kind=link}
{kind=link}
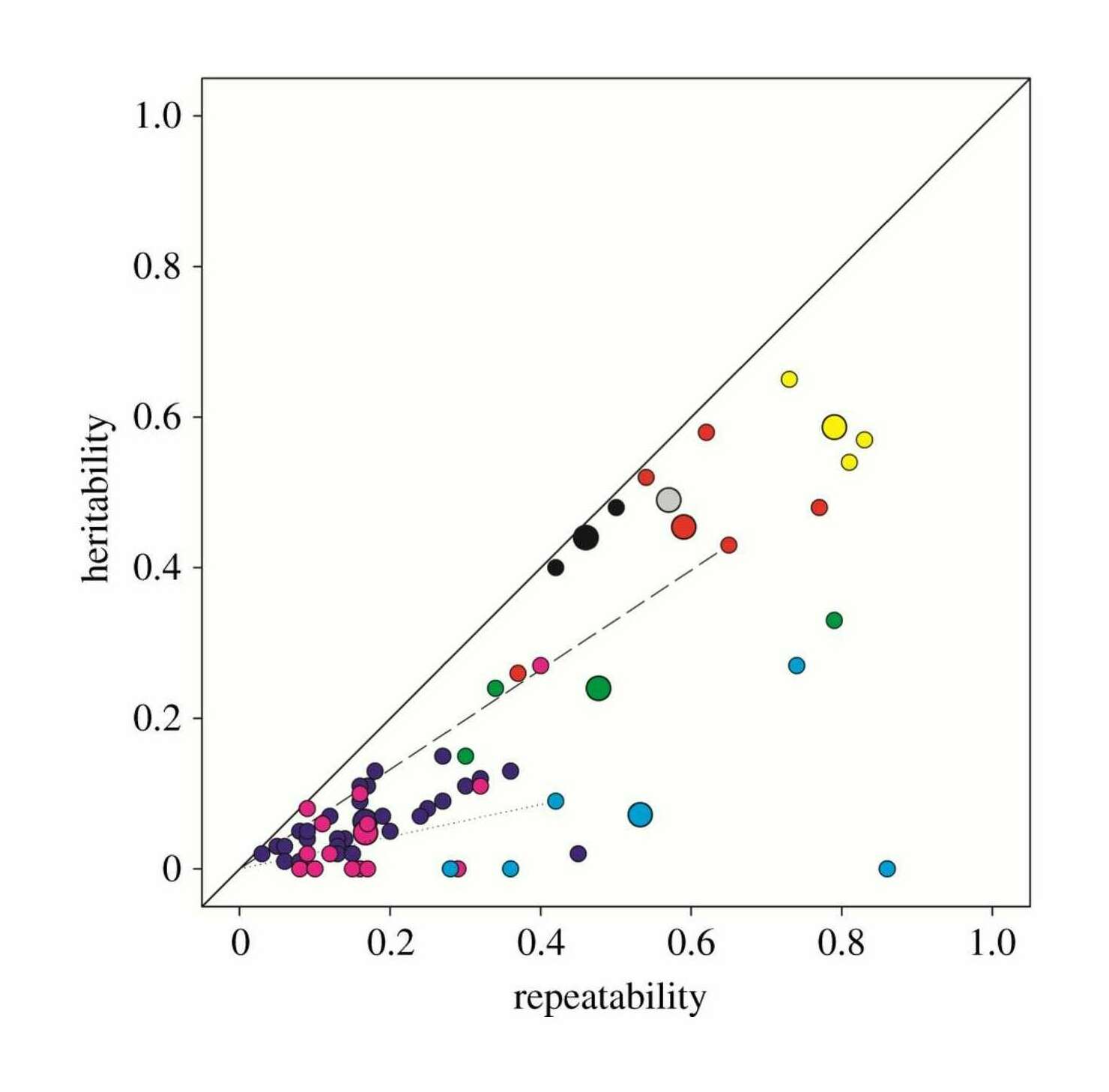{kind=link}
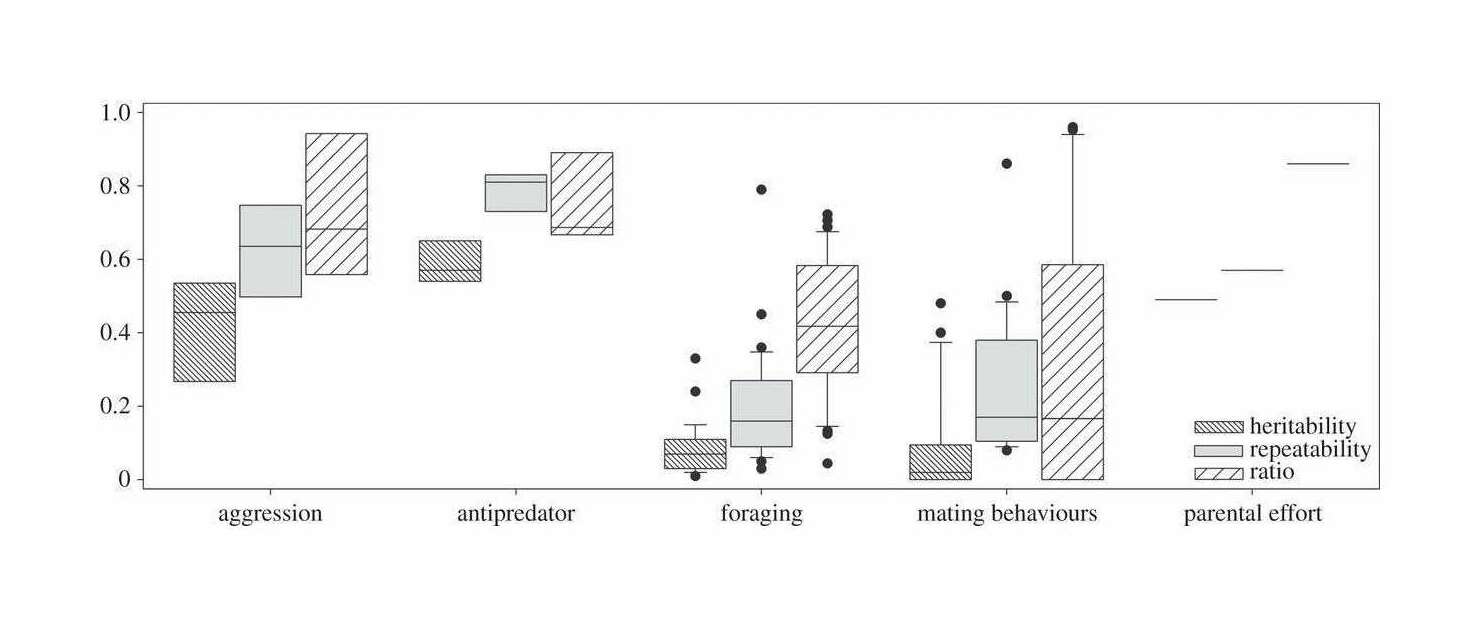{kind=link}
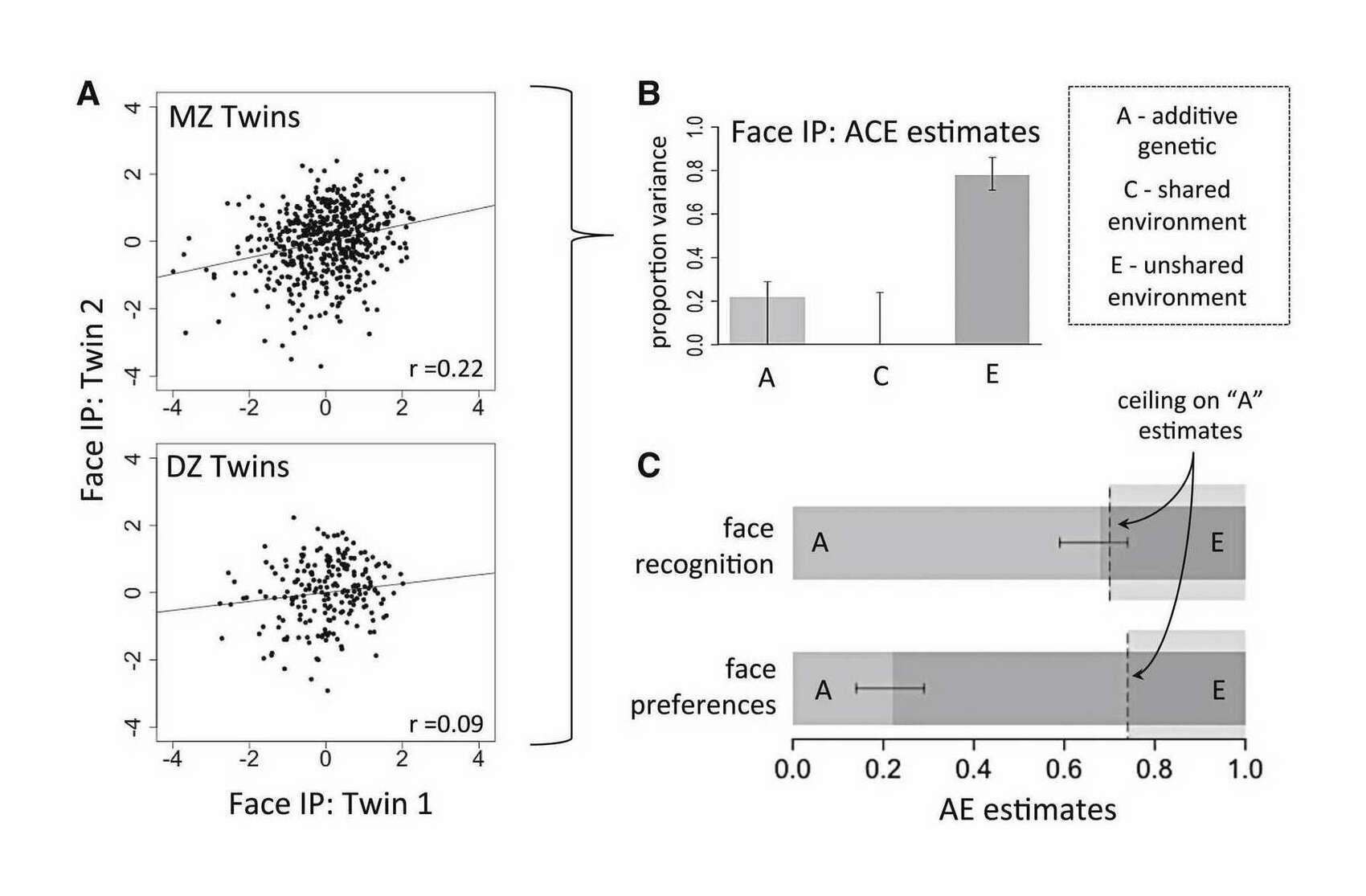{kind=link}
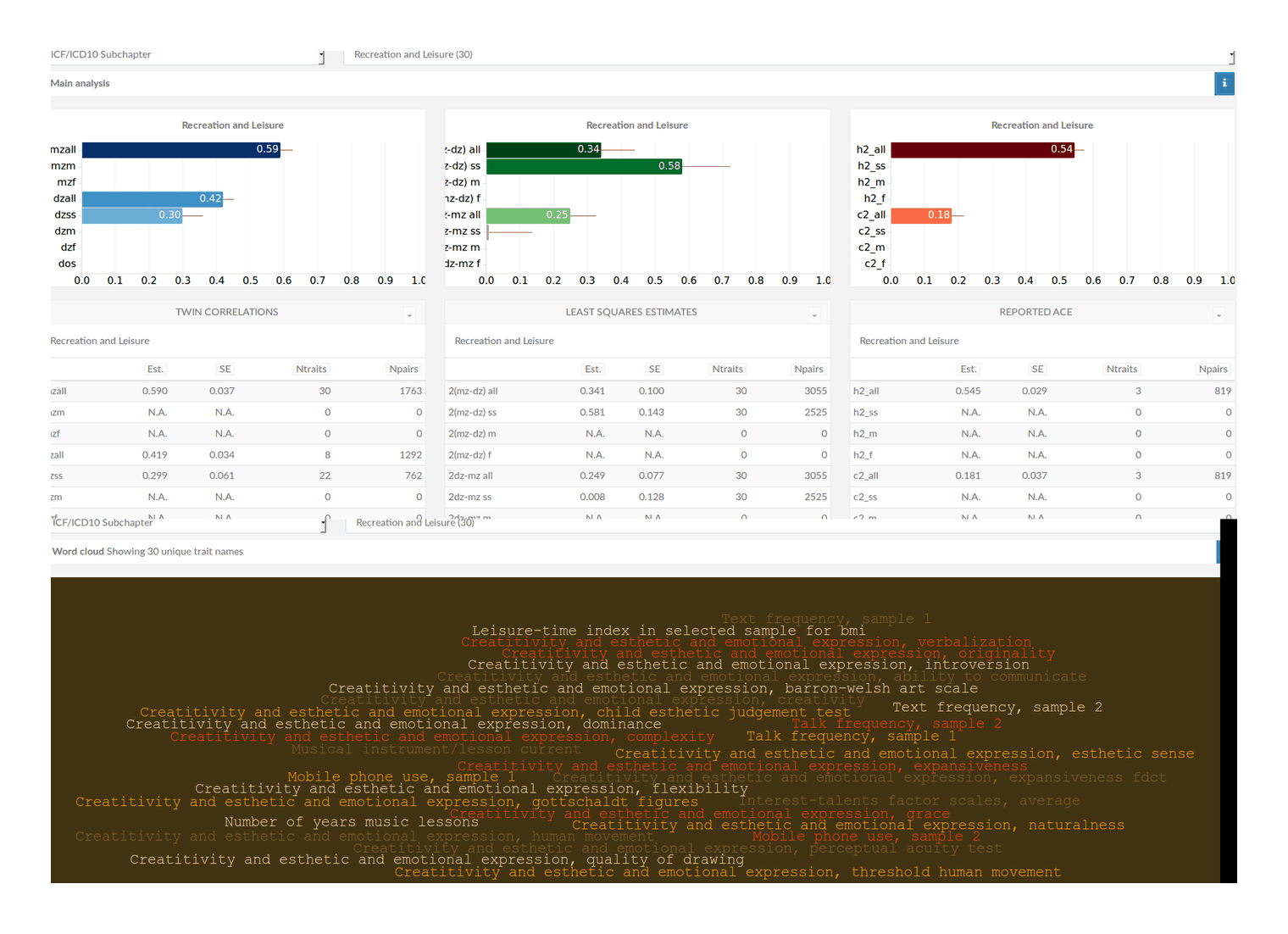{kind=link}
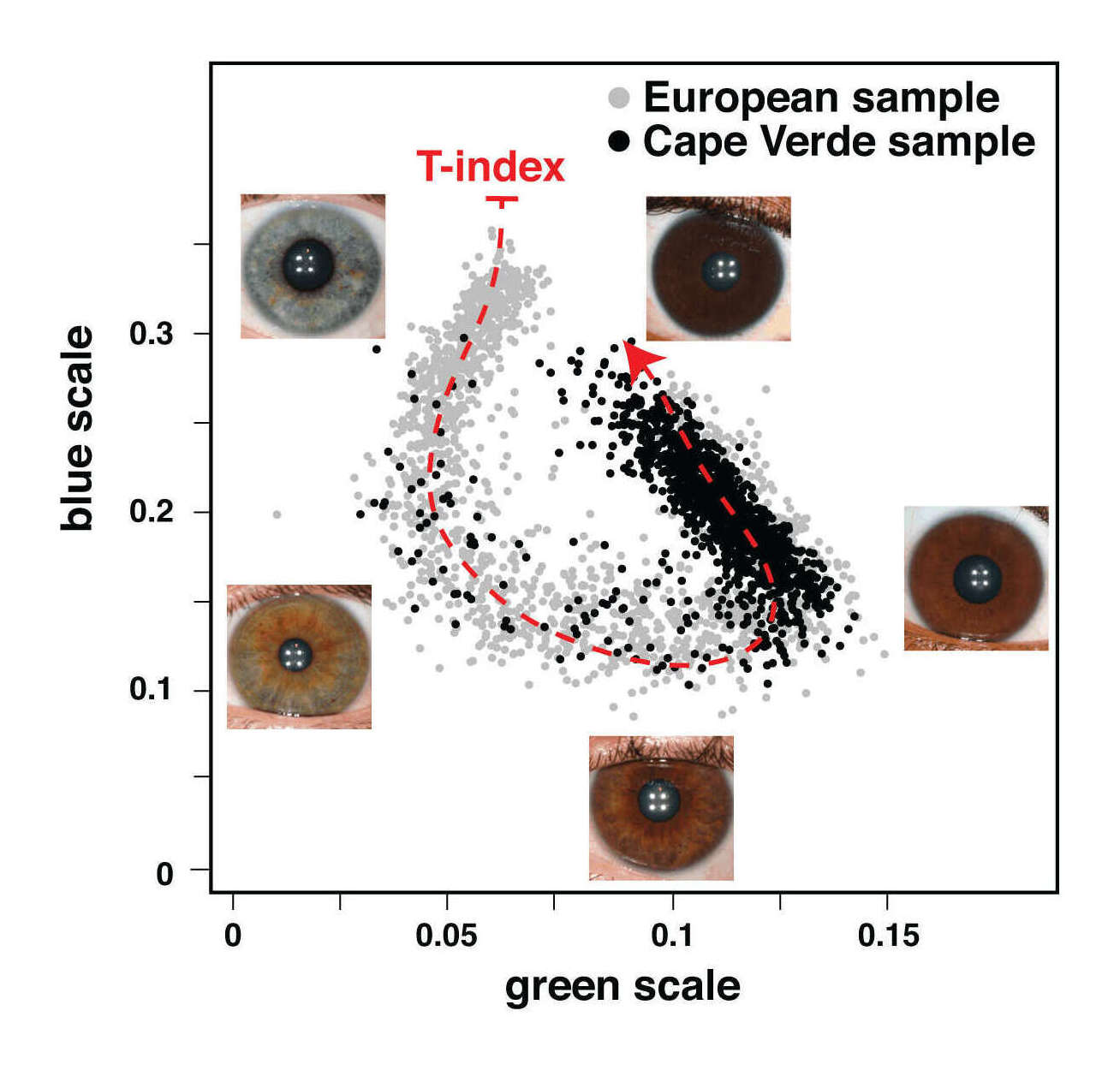{kind=link}
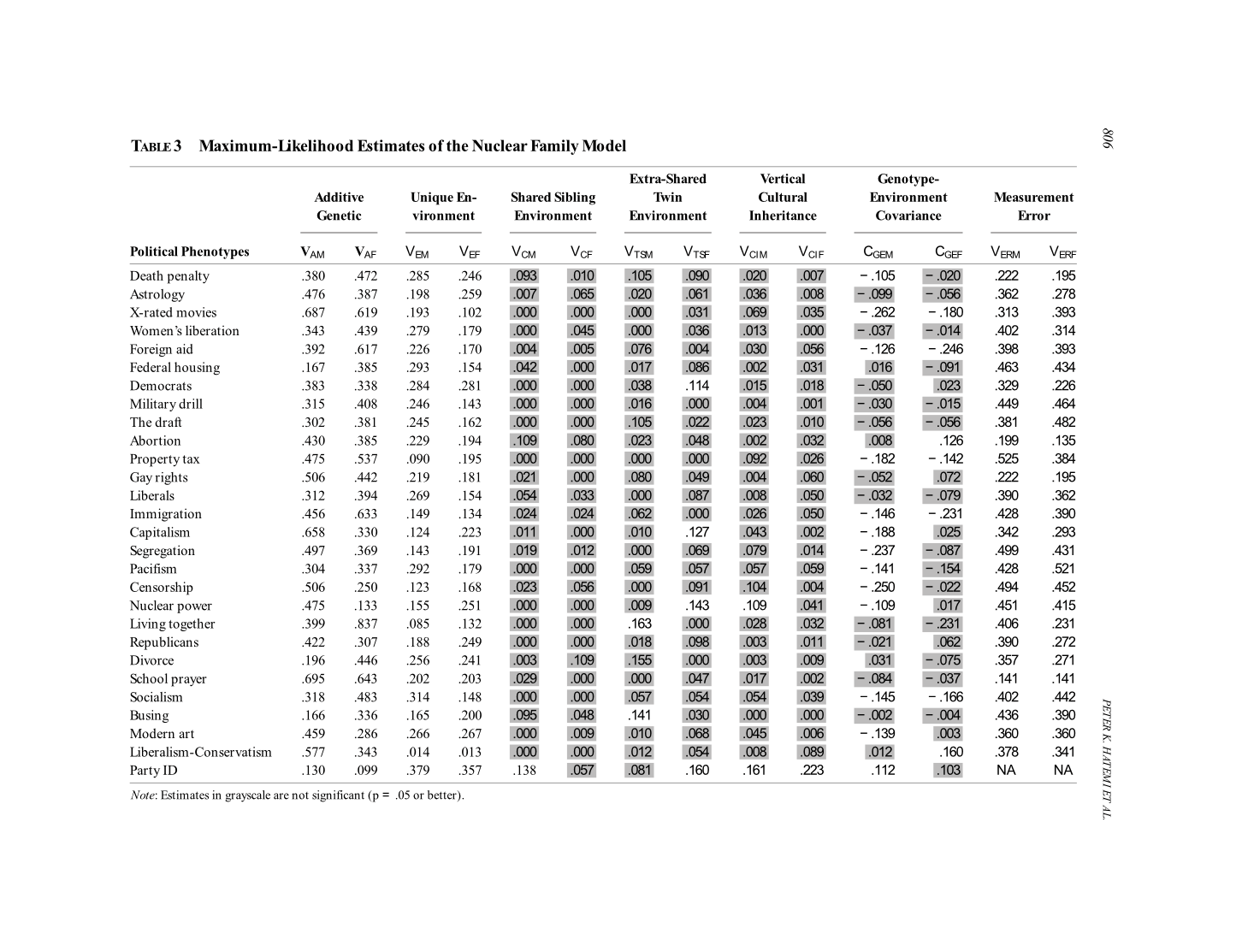{kind=link}
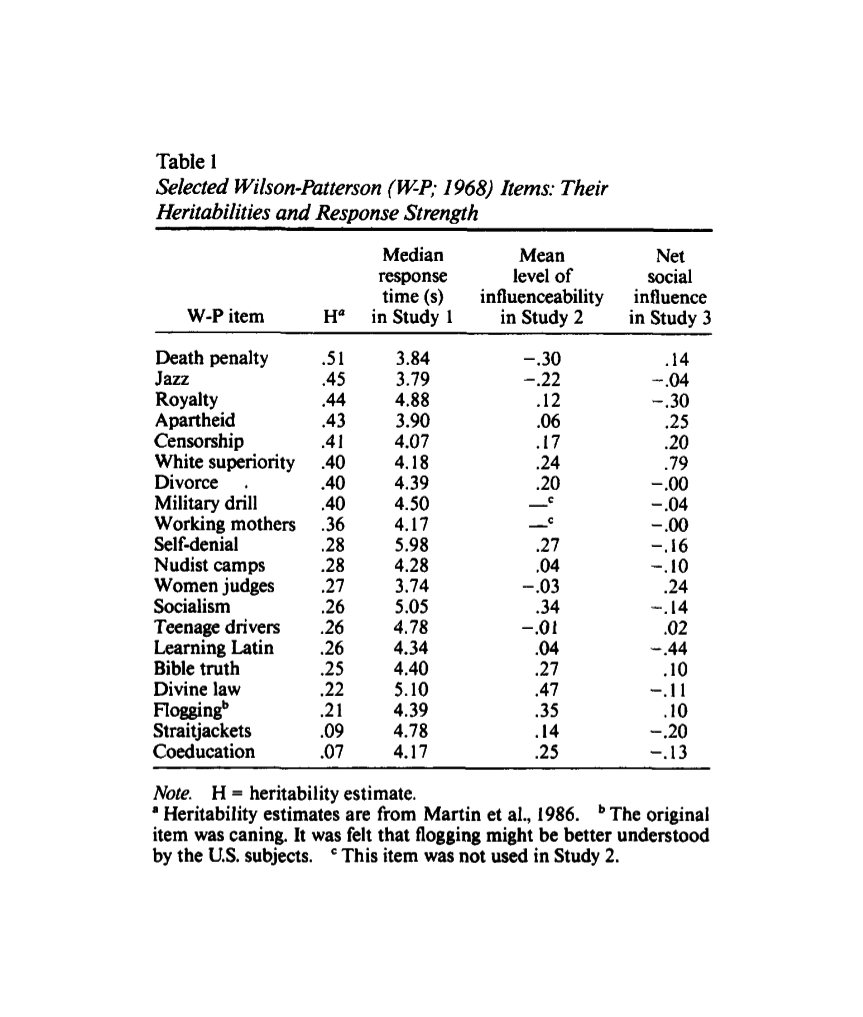{kind=link}
{kind=link}
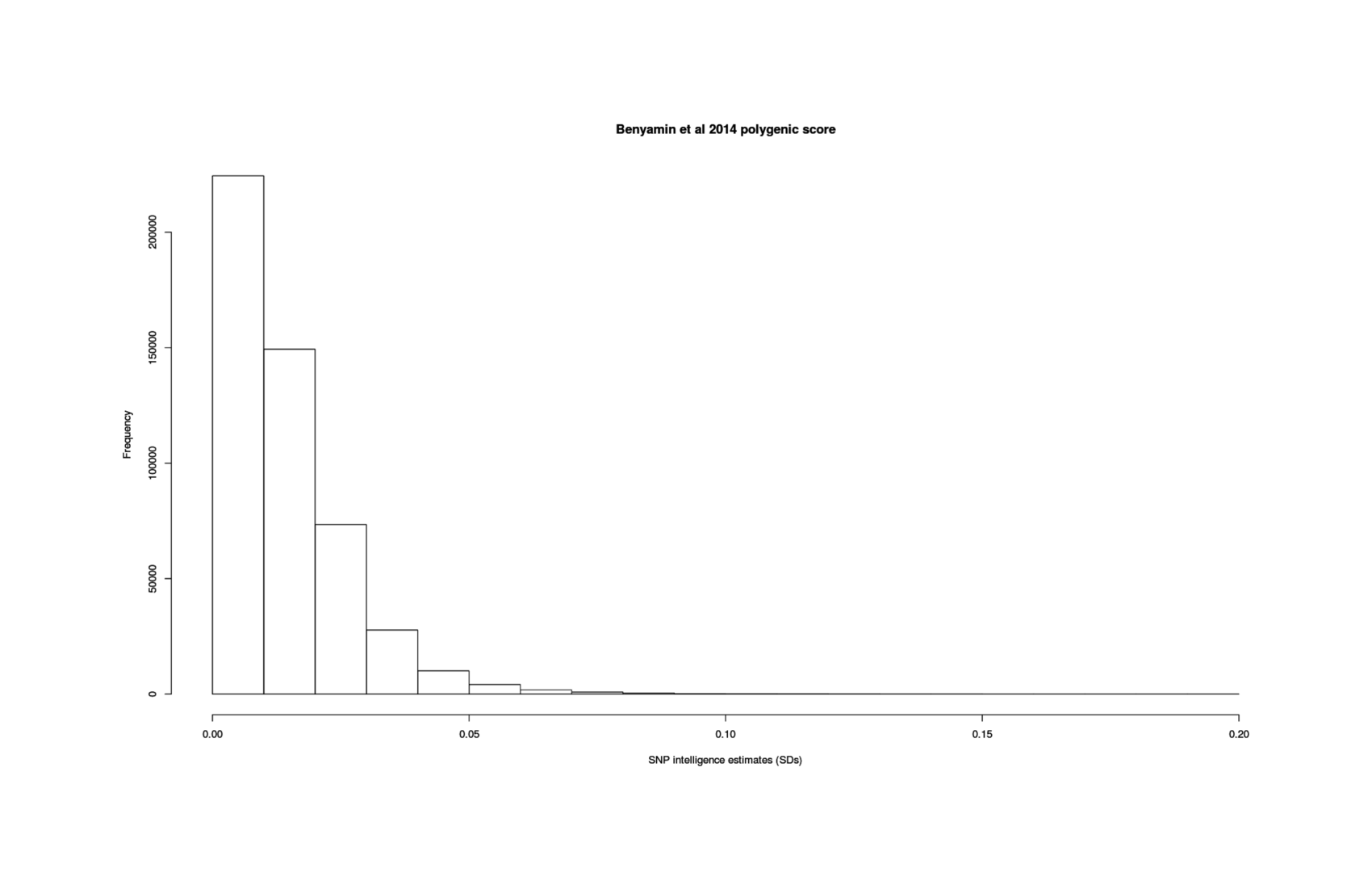{kind=link}
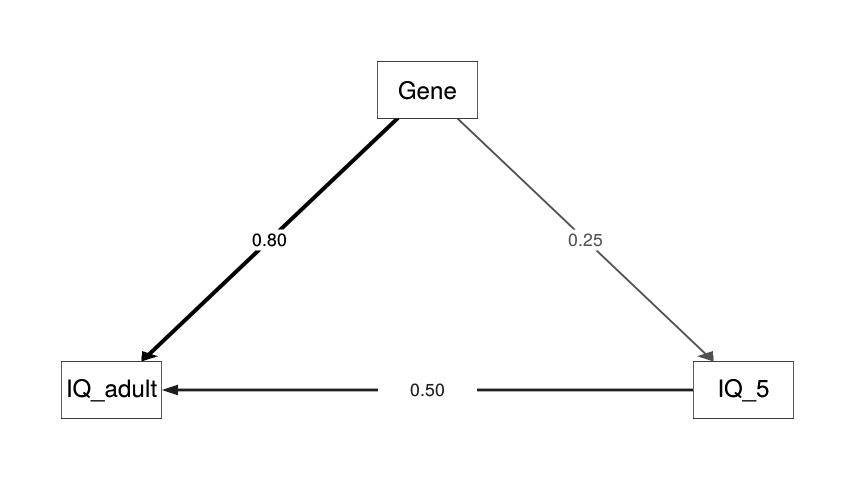{kind=link}
{kind=link}
{kind=link}
Bibliography
https://pmc.ncbi.nlm.nih.gov/articles/PMC11528883/: “A 30-Nation Investigation of Lay Heritability Beliefs”,2024-houmark.pdf: “The Nurture of Nature and the Nature of Nurture: How Genes and Investments Interact in the Formation of Skills”,https://osf.io/preprints/psyarxiv/ac7vy/: “Nature and Nurture in Fussy Eating from Toddlerhood to Early Adolescence: Findings from the Gemini Twin Cohort”,https://www.sciencedirect.com/science/article/pii/S0160289623000296: “Gene-Environment Interplay in Early Life Cognitive Development”,https://www.nature.com/articles/s42003-023-04497-8: “Sharing GWAS Summary Statistics Results in More Citations”,2023-hassan.pdf: “Genetic and Environmental Influences on Playing Video Games”,2023-abdellaoui.pdf: “15 Years of GWAS Discovery: Realizing the Promise”,2023-baldwin.pdf: “Childhood Maltreatment and Mental Health Problems: A Systematic Review and Meta-Analysis of Quasi-Experimental Studies”,2022-baldwin.pdf: “A Genetically Informed Registered Report on Adverse Childhood Experiences and Mental Health”,https://www.medrxiv.org/content/10.1101/2022.11.10.22282137.full: “Correcting for Volunteer Bias in GWAS Uncovers Novel Genetic Variants and Increases Heritability Estimates”,2022-patel.pdf: “Advances and Applications of Polygenic Scores for Coronary Artery Disease”,2022-charlesworth.pdf: “From Mendel to Quantitative Genetics in the Genome Era: the Scientific Legacy of W. G. Hill”,2022-kendler.pdf: “Medical Genetics in the 19th Century As Background to the Development of Psychiatric Genetics”,https://onlinelibrary.wiley.com/doi/full/10.1111/sms.14205: “Physical Activity and Health: Findings from Finnish Monozygotic Twin Pairs Discordant for Physical Activity”,https://onlinelibrary.wiley.com/doi/full/10.1002/gepi.22459: “Clarifying the Causes of Consistent and Inconsistent Findings in Genetics”,https://www.medrxiv.org/content/10.1101/2022.05.16.22275048.full: “Reweighting the UK Biobank to Reflect Its Underlying Sampling Population Substantially Reduces Pervasive Selection Bias due to Volunteering”,https://academic.oup.com/pnasnexus/article/1/2/pgac051/6604844: “Birth Order Differences in Education Originate in Postnatal Environments”,2022-skaug.pdf: “Childhood Trauma and Borderline Personality Disorder Traits: A Discordant Twin Study”,https://www.nature.com/articles/s41588-022-01024-z: “New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias”,https://www.sciencedirect.com/science/article/pii/S096098222101650X: “Measuring Biodiversity from DNA in the Air”,2022-makowski.pdf: “Discovery of Genomic Loci of the Human Cerebral Cortex Using Genetically Informed Brain Atlases”,2021-birmaher.pdf: “Role of Polygenic Risk Score in the Familial Transmission of Bipolar Disorder in Youth”,2021-cannonalbright.pdf: “Evidence for Excess Familial Clustering of Post Traumatic Stress Disorder in the US Veterans Genealogy Resource”,2021-giangrande.pdf: “Genetically Informed, Multilevel Analysis of the Flynn Effect across 4 Decades & 3 WISC Versions”,2021-scott.pdf: “Evolution of Sociability by Artificial Selection”,https://papers.tinbergen.nl/21088.pdf#page=3: “Using Genes to Explore the Effects of Cognitive and Non-Cognitive Skills on Education and Labor Market Outcomes”,https://www.nature.com/articles/s41398-021-01300-2: “Parental Characteristics and Offspring Mental Health and Related Outcomes: a Systematic Review of Genetically-Informative Literature”,https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1009021: “Evaluation of Polygenic Prediction Methodology within a Reference-Standardized Framework”,https://onlinelibrary.wiley.com/doi/full/10.1002/hbm.25311: “10 Years of Enhancing Neuro-Imaging Genetics through Meta-Analysis: An Overview from the ENIGMA Genetics Working Group”,https://www.science.org/content/article/landmark-study-resolves-major-mystery-how-genes-govern-human-height: “‘Landmark’ Study Resolves a Major Mystery of How Genes Govern Human Height”,2019-hyytinen.pdf: “Heritability of Lifetime Earnings”,2018-roselli.pdf: “Multi-Ethnic Genome-Wide Association Study for Atrial Fibrillation”,2018-jamnik.pdf: “A Multimethodological Study of Preschoolers’ Preferences for Aggressive Television and Video Games”,2018-claes.pdf: “Genome-Wide Mapping of Global-To-Local Genetic Effects on Human Facial Shape”,2017-schmidt.pdf: “Beyond Questionable Research Methods: The Role of Omitted Relevant Research in the Credibility of Research”,https://www.cell.com/current-biology/fulltext/S0960-9822(15)01019-2: “Individual Esthetic Preferences for Faces Are Shaped Mostly by Environments, Not Genes”,2015-dezeeuw.pdf: “Meta-Analysis of Twin Studies Highlights the Importance of Genetic Variation in Primary School Educational Achievement”,2015-nelson.pdf: “The Support of Human Genetic Evidence for Approved Drug Indications”,https://royalsocietypublishing.org/doi/full/10.1098/rspb.2014.2201: “The Contribution of Additive Genetic Variation to Personality Variation: Heritability of Personality”,2013-conley.pdf: “Heritability and the Equal Environments Assumption: Evidence from Multiple Samples of Misclassified Twins”,2008-visscher.pdf: “Heritability in the Genomics Era—Concepts and Misconceptions”,2006-segal.pdf: “Two Monozygotic Twin Pairs Discordant for Female-To-Male Transsexualism”,2003-jacob.pdf: “Genetic and Environmental Effects on Offspring Alcoholism: New Insights Using an Offspring-Of-Twins Design”,2001-dickens.pdf: “Heritability Estimates versus Large Environmental Effects: The IQ Paradox Resolved”,https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(99)90364-1/fulltext: “Early Observations of Genetic Diseases”,