- See Also
-
Links
- “Design of Highly Functional Genome Editors by Modeling the Universe of CRISPR-Cas Sequences”, Ruffolo et al 2024
- “AAV1-HOTOF Gene Therapy for Autosomal Recessive Deafness 9: a Single-Arm Trial”, Lv et al 2024
- “Cryptography in the DNA of Living Cells Enabled by Multi-Site Base Editing”, Volf et al 2023
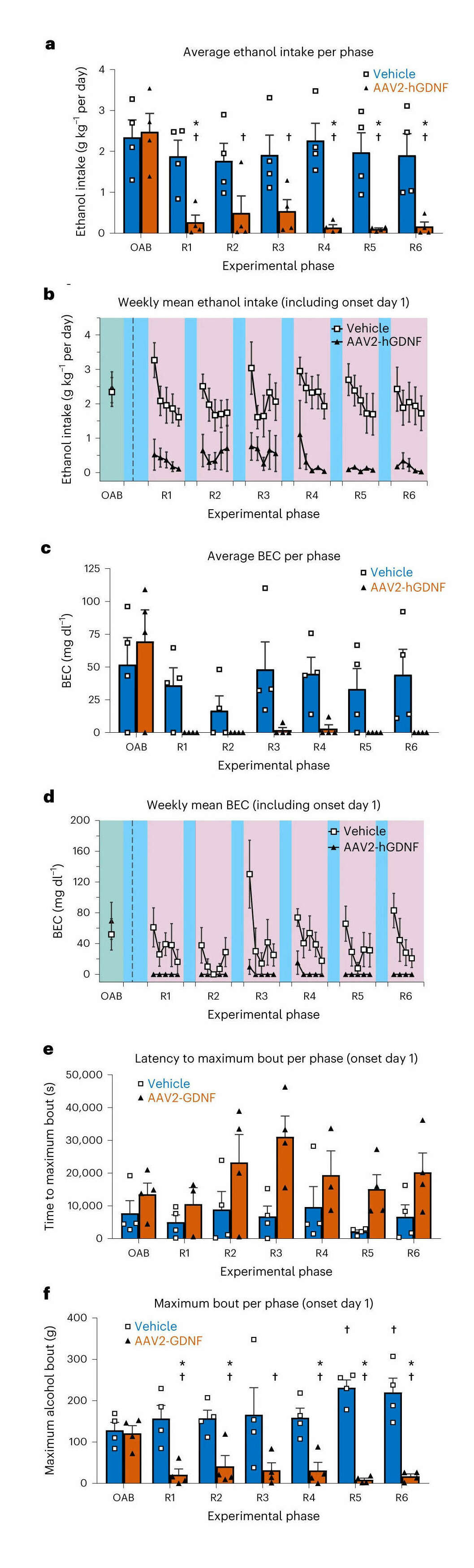
- “GDNF Gene Therapy for Alcohol Use Disorder in Male Non-Human Primates”, Ford et al 2023b
- “In Vivo Biosynthesis of N,N-Dimethyltryptamine, 5-MeO-N,N-Dimethyltryptamine, and Bufotenine in E. Coli”, Friedberg et al 2023
- “Temperature-Dependent RNA Editing in Octopus Extensively Recodes the Neural Proteome”, Birk et al 2023
- “National and Global Impacts of Genetically Modified Crops”, Hansen & Wingender 2023
- “The First Crispr-Edited Salad Is Here: A Startup Used Gene Editing to Make Mustard Greens More Appetizing to Consumers. Next Up: Fruits”, Mullin 2023
- “First Gene-Edited Calf With Reduced Susceptibility to a Major Viral Pathogen”, Workman et al 2023
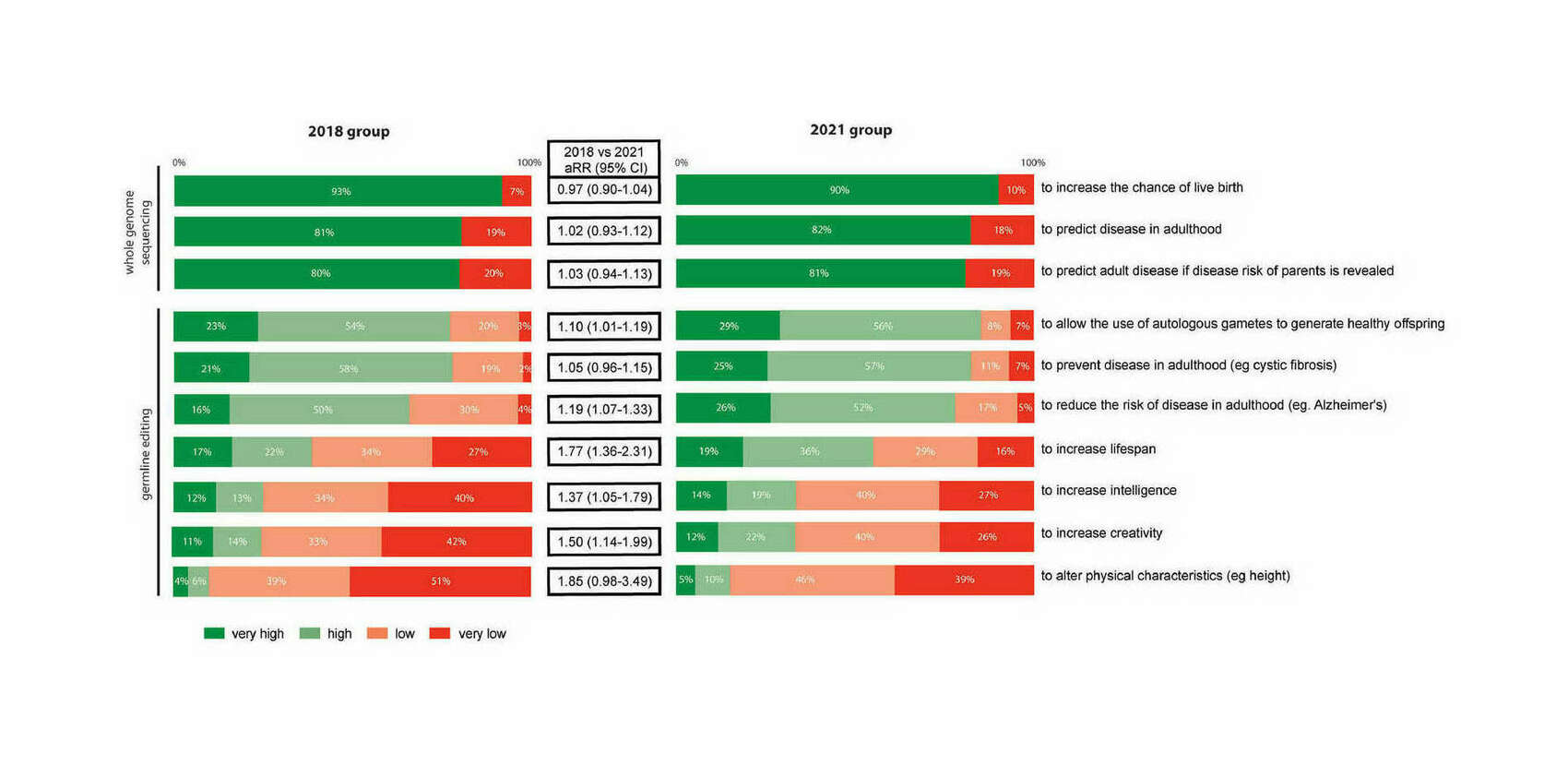
- “Acceptance of Genetic Editing and of Whole Genome Sequencing of Human Embryos by Patients With Infertility Before and After the Onset of the COVID-19 Pandemic”, Neuhausser et al 2023
- “CRISPR Technology: A Decade of Genome Editing Is Only the Beginning”, Wang & Doudna 2023
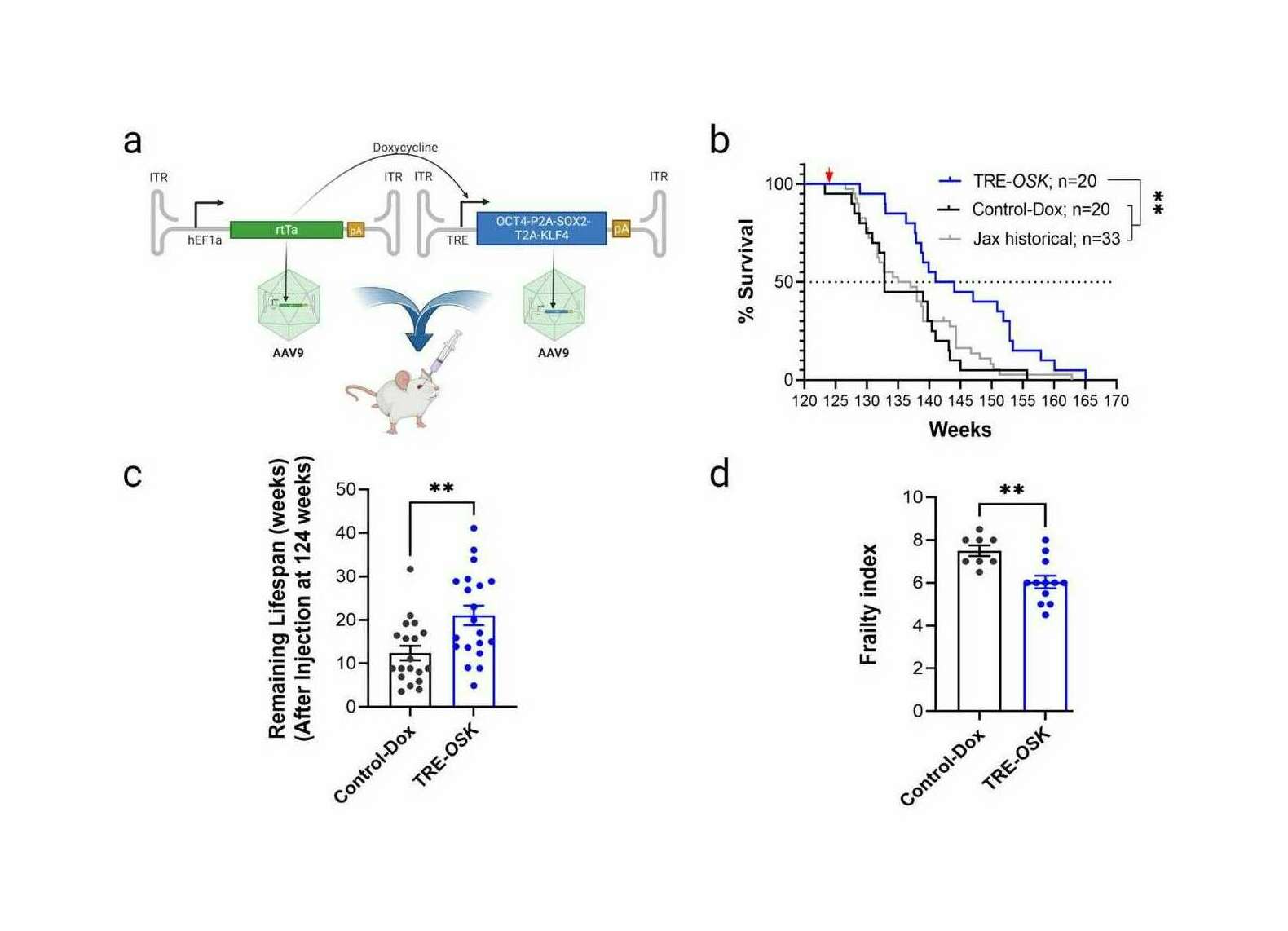
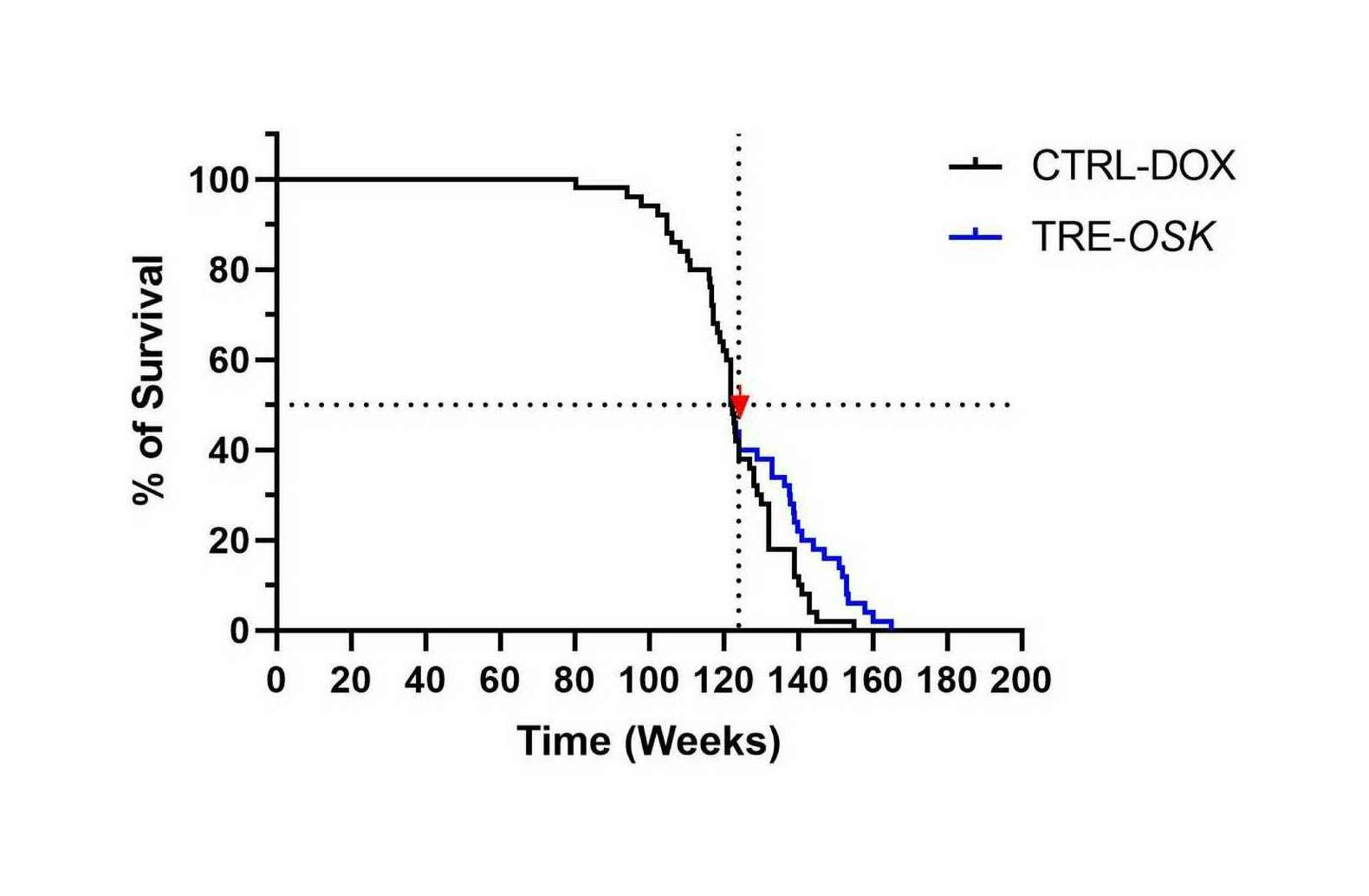
- “Gene Therapy Mediated Partial Reprogramming Extends Lifespan and Reverses Age-Related Changes in Aged Mice”, Macip et al 2023
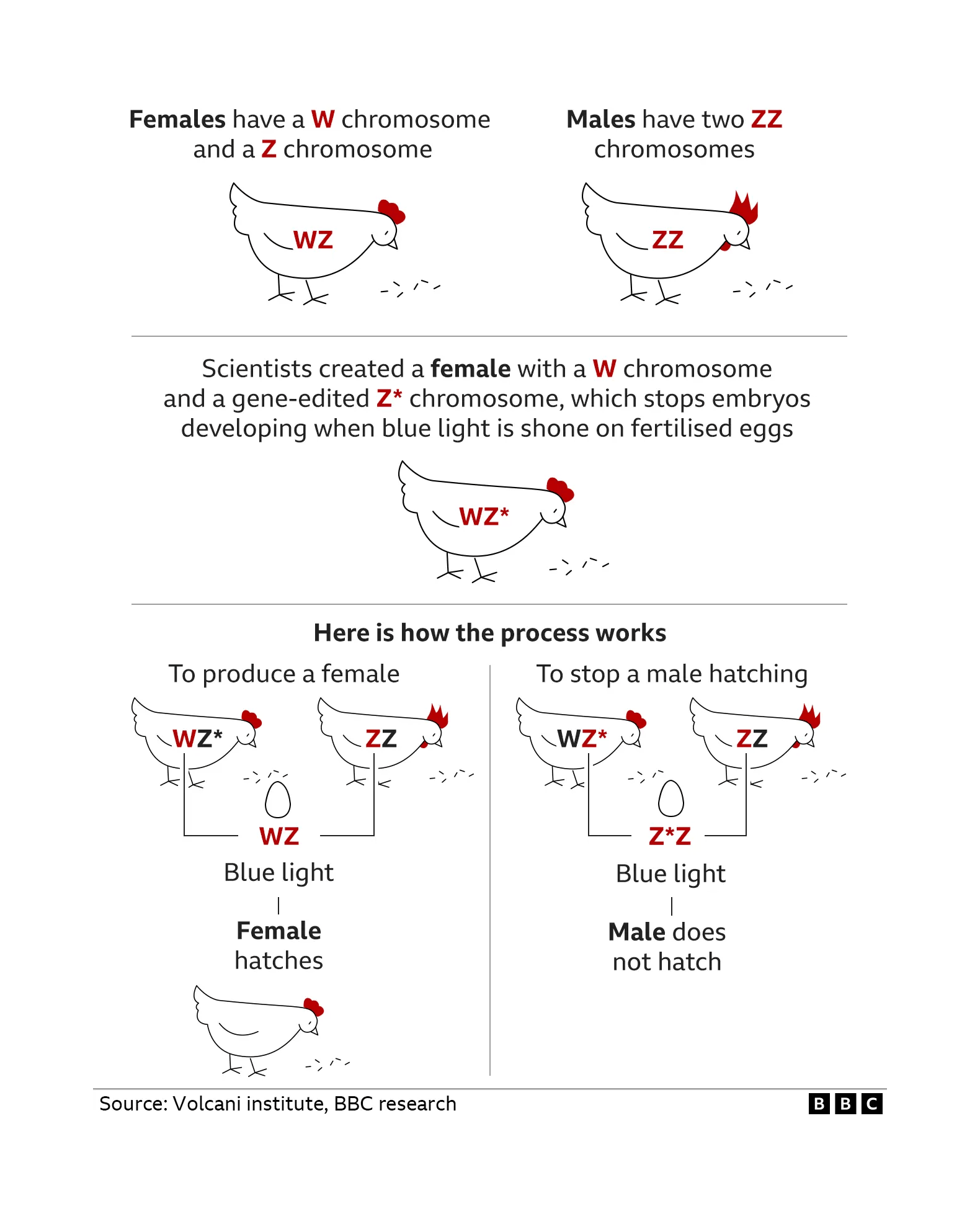
- “Gene-Edited Hens May End Cull of Billions of Chicks”, Ghosh 2022
- “Base Editing: Revolutionary Therapy Clears Girl’s Incurable Cancer”, Gallagher 2022
- “MicroRNAs Are Deeply Linked to the Emergence of the Complex Octopus Brain”, Zolotarov et al 2022
- “PASTE: Drag-And-Drop Genome Insertion of Large Sequences without Double-Strand DNA Cleavage Using CRISPR-Directed Integrases”, Yarnall et al 2022
- “Large Scale Functional Screen Identifies Genetic Variants With Splicing Effects in Modern and Archaic Humans”, Rong et al 2022
- “Prime Editing for Precise and Highly Versatile Genome Manipulation”, Chen & Liu 2022b
- “Role of Spike in the Pathogenic and Antigenic Behavior of SARS-CoV-2 BA.1 Omicron”, Chen et al 2022
- “New Self-Sexing Aedes Aegypti Strain Eliminates Barriers to Scalable and Sustainable Vector Control for Governments and Communities in Dengue-Prone Environments”, Spinner et al 2022
- “RNA Recoding in Cephalopods Tailors Microtubule Motor Protein Function”, Rangan & Reck-Peterson 2022
- “A Sustainable Mouse Karyotype Created by Programmed Chromosome Fusion”, Wang et al 2022e
- “CRISPR-Cas9-Mediated Gene Editing of the BCL11A Enhancer for Pediatric Β0/β0 Transfusion-Dependent Β-Thalassemia”, Fu et al 2022
- “A Versatile, High-Efficiency Platform for CRISPR-Based Gene Activation”, Heidersbach et al 2022
- “Rubisco Function, Evolution, and Engineering”, Prywes et al 2022
- “Generation of Genome-Edited Dogs by Somatic Cell Nuclear Transfer”, Kim et al 2022
- “Therapeutic in Vivo Delivery of Gene Editing Agents”, Raguram et al 2022
- “Retro-Cascorder: Recording Gene Expression Order in DNA by CRISPR Addition of Retron Barcodes”, Bhattarai-Kline et al 2022
- “Genome-Edited Crops for Improved Food Security of Smallholder Farmers”, Pixley et al 2022
- “The Creator of the CRISPR Babies Has Been Released from a Chinese Prison: He Jiankui Created the First Gene-Edited Children. The Price Was His Career. And His Freedom.”, Regalado 2022
- “A CRISPR Kitty? Gene Editing Breathes New Life into the Hypoallergenic Cat”, Lin 2022
- “Virgin Birth: A Genetic Basis for Facultative Parthenogenesis”, Braun et al 2022
- “Viable Offspring Derived from Single Unfertilized Mammalian Oocytes”, Wei et al 2022b
- “How to Protect the First ‘CRISPR Babies’ Prompts Ethical Debate: Fears of Excessive Interference Cloud Proposal for Protecting Children Whose Genomes Were Edited, As He Jiankui’s Release from Jail Looks Imminent”, Mallapaty 2022
- “In Vivo Mitochondrial Base Editing via Adeno-Associated Viral Delivery to Mouse Post-Mitotic Tissue”, Silva-Pinheiro et al 2022
- “The Gene TaWOX5 Overcomes Genotype Dependency in Wheat Genetic Transformation”, Wang et al 2022
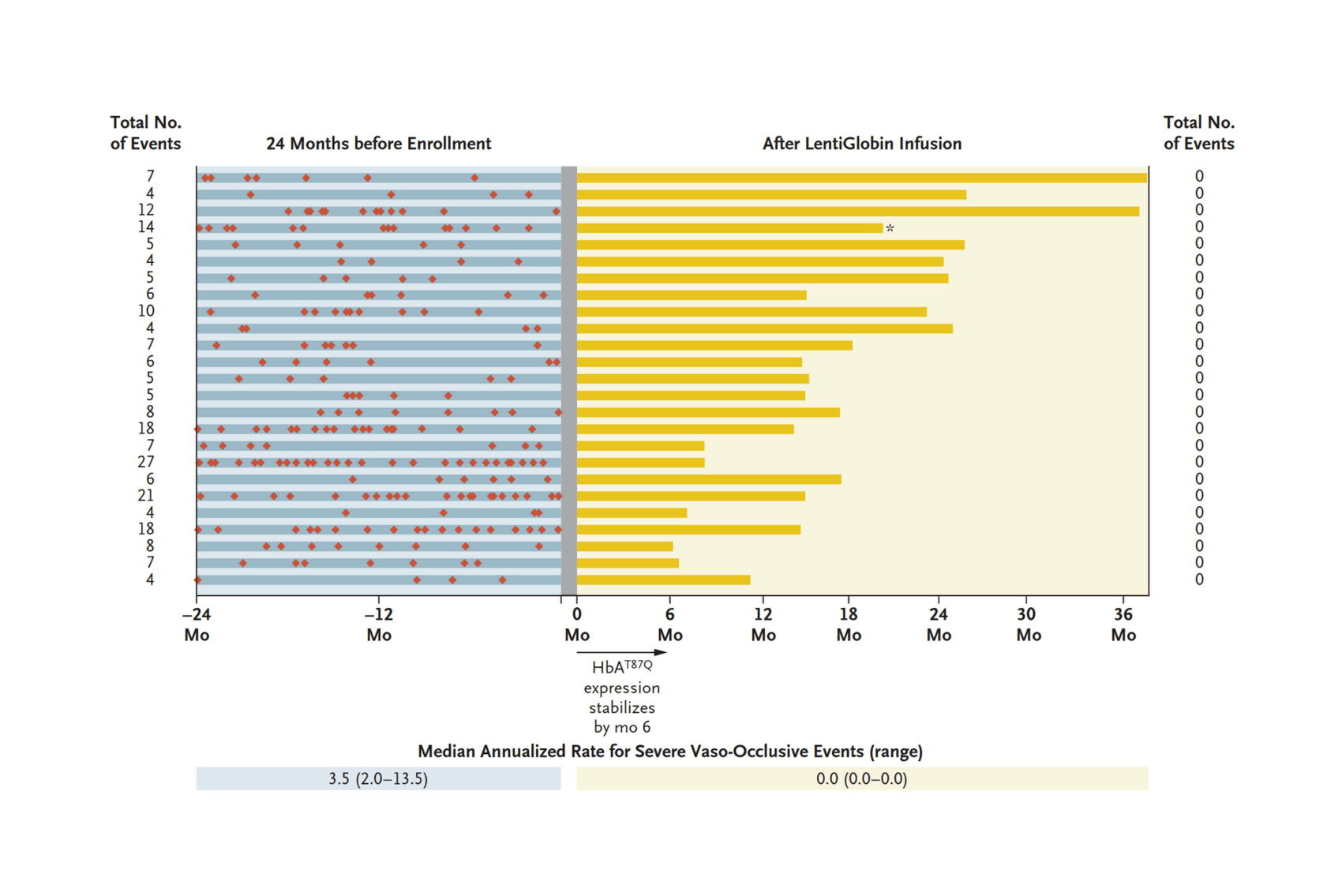
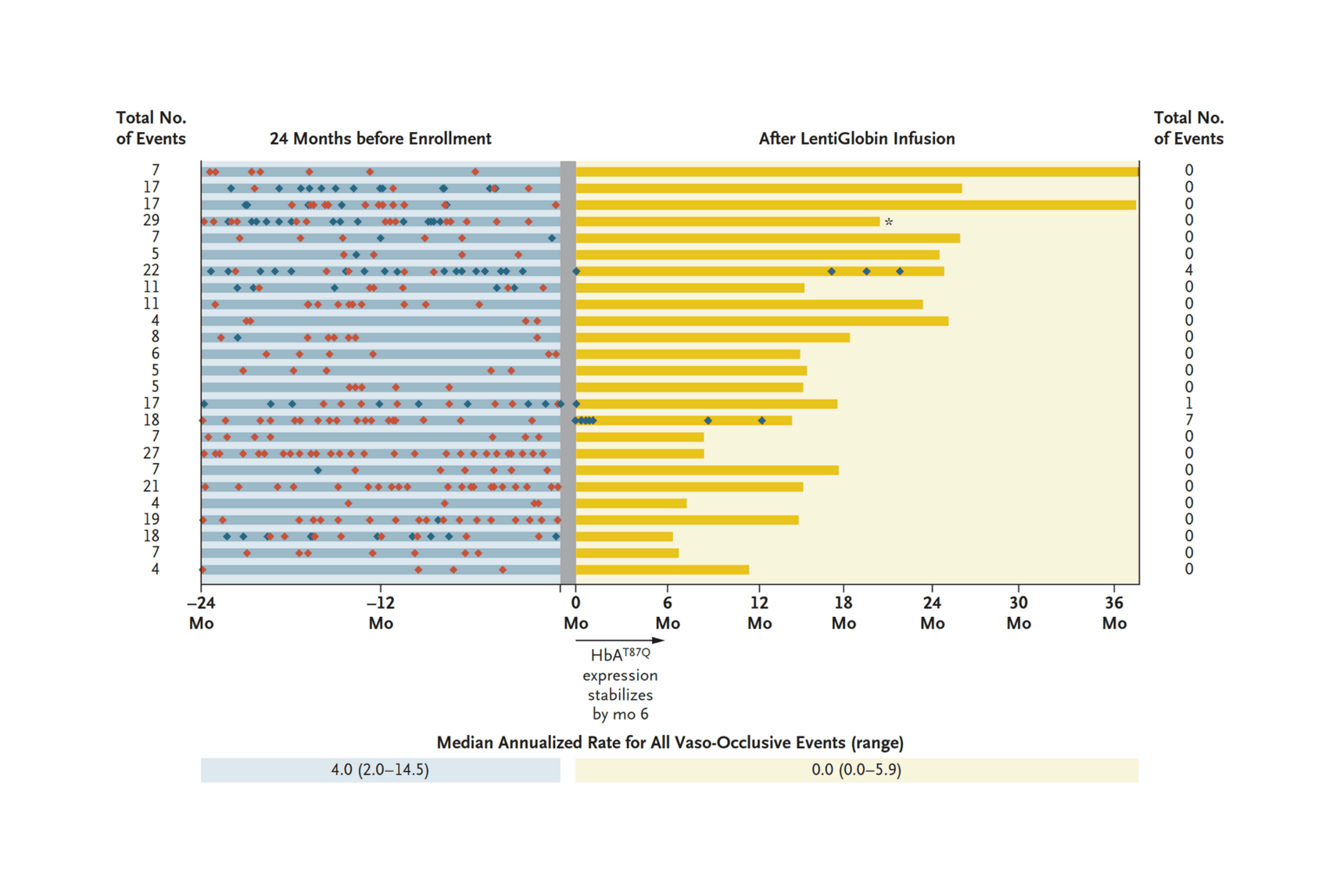
- “Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease”, Kanter et al 2021
- “CRISPR-Cas9 Effectors Facilitate Generation of Single-Sex Litters and Sex-Specific Phenotypes”, Douglas et al 2021
- “Lipid Nanoparticles Incorporating a GalNAc Ligand Enable in Vivo Liver ANGPTL3 Editing in Wild-Type and Somatic LDLR Knockout Non-Human Primates”, Kasiewicz et al 2021
- “A Temporally Resolved, Multiplex Molecular Recorder Based on Sequential Genome Editing”, Choi et al 2021
- “Multiplex Genomic Recording of Enhancer and Signal Transduction Activity in Mammalian Cells”, Chen et al 2021
- “Drag-And-Drop Genome Insertion without DNA Cleavage With CRISPR-Directed Integrases”, Ioannidi et al 2021
- “PRIME-Del: Precise Genomic Deletions Using Paired Prime Editing”, Choi et al 2021
- “PEDAR: Deletion and Replacement of Long Genomic Sequences Using Prime Editing”, Jiang et al 2021
- “In a First, Surgeons Attached a Pig Kidney to a Human, and It Worked: A Kidney Grown in a Genetically Altered Pig Functions Normally, Scientists Reported. The Procedure May Open the Door to a Renewable Source of Desperately Needed Organs”, Rabin 2021
- “The Widespread IS200/605 Transposon Family Encodes Diverse Programmable RNA-Guided Endonucleases”, Altae-Tran et al 2021
- “Mammalian Retrovirus-Like Protein PEG10 Packages Its Own MRNA and Can Be Pseudotyped for MRNA Delivery”, Segel et al 2021
- “Gene-Drive Suppression of Mosquito Populations in Large Cages As a Bridge between Lab and Field”, Hammond et al 2021
- “RNA Demethylation Increases the Yield and Biomass of Rice and Potato Plants in Field Trials”, Yu et al 2021b
- “Surrogate Broodstock to Enhance Biotechnology Research and Applications in Aquaculture”, Jin et al 2021
- “CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis”, Gillmore et al 2021
- “Base Editing of Haematopoietic Stem Cells Rescues Sickle Cell Disease in Mice”, Newby et al 2021
- “Scientists Drove Mice to Bond by Zapping Their Brains With Light: The Study, a Tour De Force in Bioengineering, Comes After Two Decades of Research on Brain-To-Brain Synchrony in People”, Hughes 2021
- “Partial Recovery of Visual Function in a Blind Patient After Optogenetic Therapy”, Sahel et al 2021
- “With Engineered Proteins, Scientists Use Optogenetics for the First Time to Help a Blind Patient See Again”, Molteni 2021
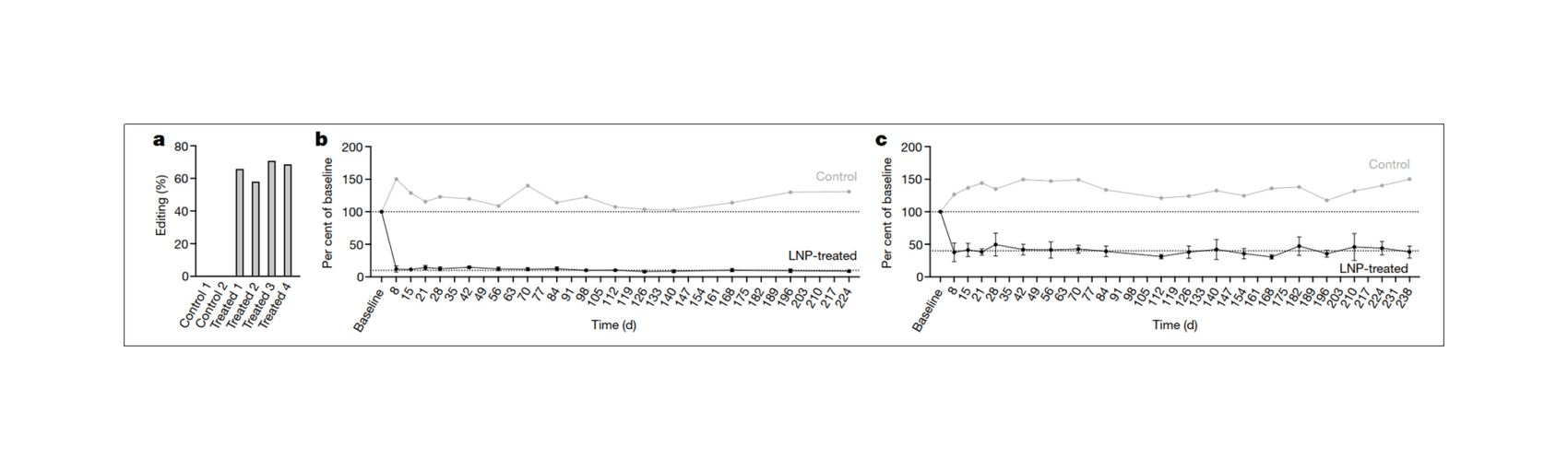
- “In Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates”, Musunuru et al 2021
- “High-Throughput Functional Variant Screens via in Vivo Production of Single-Stranded DNA”, Schubert et al 2021
- “First Genetically Modified Mosquitoes Released in the United States: Biotech Firm Oxitec Launches Controversial Field Test of Its Insects in Florida After Years of Push-Back from Residents and Regulatory Complications”, Waltz 2021
- “Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing”, Nuñez et al 2021
- “China Officially Bans CRISPR Babies, Human Clones and Animal-Human Hybrids”, News 2021
- “Reintroduction of the Archaic Variant of NOVA1 in Cortical Organoids Alters Neurodevelopment”, Trujillo et al 2021
- “Reactivation of the Pluripotency Program Precedes Formation of the Cranial Neural Crest”, Zalc et al 2021
- “In Vivo Base Editing Rescues Hutchinson-Gilford Progeria Syndrome in Mice”, Koblan et al 2021
- “Expression of Functional Plant Sweet Protein Thaumatin II in the Milk of Transgenic Mice”, Lu et al 2021
- “Xenogeneic Stem Cell Transplantation: Research Progress and Clinical Prospects”, Jiang et al 2021
- “Human-Animal Interspecies Chimerism via Blastocyst Complementation: Advances, Challenges and Perspectives: a Narrative Review”, Li & Huang 2021
- “Reconstitution of the Oocyte Transcriptional Network With Transcription Factors”, Hamazaki et al 2020
- “Biotechnology Research Viewed With Caution Globally, but Most Support Gene Editing for Babies To Treat Disease: Majorities across Global Publics Accept Evolution; Religion Factors Prominently in Belief”, Funk et al 2020
- “Reprogramming to Recover Youthful Epigenetic Information and Restore Vision”, Lu et al 2020
- “Inclusion of Variants Discovered from Diverse Populations Improves Polygenic Risk Score Transferability”, Cavazos & Witte 2020
- “Recency Negativity: Newer Food Crops Are Evaluated Less Favorably”, Inbar et al 2020
- “CRISPR-Enhanced Human Adipocyte ‘Browning’ As Cell Therapy for Metabolic Disease”, Tsagkaraki et al 2020
- “Press Release: The Nobel Prize in Chemistry 2020”, Sciences 2020
- “Heritable Human Genome Editing”, Report 2020
- “Human Embryo Gene Editing Gets a Road Map—Not a Green Light: After the 2018 ‘Crispr Baby’ Scandal, a Global Commission Assessed the Technology and Set Strict Criteria for Moving It toward Clinical Trials”, Molteni 2020
- “CRISPR-Engineered Human Brown-Like Adipocytes Prevent Diet-Induced Obesity and Ameliorate Metabolic Syndrome in Mice”, Wang et al 2020d
- “An Antiviral Self-Replicating Molecular Heterotroph”, Shapiro et al 2020
- “Human-Specific ARHGAP11B Increases Size and Folding of Primate Neocortex in the Fetal Marmoset”, Heide et al 2020
- “Cas9 Cuts and Consequences; Detecting, Predicting, and Mitigating CRISPR/Cas9 On-Target and Off-Target Damage [Techniques for Detecting, Predicting, and Mitigating the On-Target and Off-Target Effects of Cas9 Editing]”, Newman et al 2020
- “A Bacterial Cytidine Deaminase Toxin Enables CRISPR-Free Mitochondrial Base Editing”, Mok et al 2020
- “A Year In, 1st Patient To Get Gene Editing For Sickle Cell Disease Is Thriving”, Stein 2020
- “Three People With Inherited Diseases Successfully Treated With CRISPR”, Page 2020
- “CRISPR-Cas9-Mediated Induction of Heritable Chromosomal Translocations in Arabidopsis”, Beying et al 2020
- “Reversal of Aging via in Vivo Epigenetic Reprogramming”, Lu 2020b
- “Generation of Human Endothelium in Pig Embryos Deficient in ETV2”, Das et al 2020
- “The Promise and Challenge of Therapeutic Genome Editing”, Doudna 2020
- “Metabolic Engineering of Saccharomyces Cerevisiae for the de Novo Production of Psilocybin and Related Tryptamine Derivatives”, Milne et al 2020
- “Chinese Scientist Who Genetically Edited Babies Gets 3 Years in Prison: He Jiankui’s Work Was Also Carried out on a Third Infant, according to China’s State Media, in a New Disclosure That Is Likely to Add to the Global Uproar over Such Experiments.”, Wee 2019
- “Extensive Mammalian Germline Genome Engineering”, Yue et al 2019
- “Eyeing Organs for Human Transplants, Companies Unveil the Most Extensively Gene-Edited Pigs Yet”, Servick 2019
- “A Single Combination Gene Therapy Treats Multiple Age-Related Diseases”, Davidsohn et al 2019
- “Search-And-Replace Genome Editing without Double-Strand Breaks or Donor DNA”, Anzalone et al 2019
- “New ‘Prime’ Genome Editor Could Surpass CRISPR”, Cohen 2019
- “A New Crispr Technique Could Fix Almost All Genetic Diseases: A Less Error-Prone DNA Editing Method Could Correct Many More Harmful Mutations Than Was Previously Possible”, Molteni 2019
- “Technological Challenges and Milestones for Writing Genomes: Synthetic Genomics Requires Improved Technologies”, Ostrov et al 2019
- “CRISPR-Edited Stem Cells in a Patient With HIV and Acute Lymphocytic Leukemia”, Xu et al 2019
- “The Promise and Price of Cellular Therapies: New ‘Living Drugs’—Made from a Patient’s Own Cells—Can Cure Once Incurable Cancers. But Can We Afford Them?”, Mukherjee 2019
- “Russian Biologist Plans More CRISPR-Edited Babies: The Proposal Follows a Chinese Scientist Who Claimed to Have Created Twins from Edited Embryos Last Year”, Cyranoski 2019
- “Exclusive: 5 Couples Lined up for CRISPR Babies to Avoid Deafness”, Page 2019
- “Amid Animal Cruelty Debate, 80% of South Korea’s Sniffer Dogs Are Cloned”, Tribune 2019
- “When Genome Editing Goes Off-Target: Detecting Unintended Mutations Could Improve DNA-Editing Strategies”, Kempton & Qi 2019
- “Enabling Large-Scale Genome Editing by Reducing DNA Nicking”, Smith et al 2019
- “Super-Mendelian Inheritance Mediated by CRISPR-Cas9 in the Female Mouse Germline”, Grunwald et al 2019
- “The Transferability of Lipid-Associated Loci across African, Asian and European Cohorts”, Telkar et al 2019
- “Improving Crop Yield: Synthetic Photorespiration Bypass Increases Crop Yield”, Eisenhut & Weber 2019
- “Molecular Digital Data Storage Using DNA”, Ceze et al 2019
- “After the Storm—A Responsible Path for Genome Editing”, Daley et al 2019
- “Is the ‘SErious’ Factor in Germline Modification Really Relevant? A Response to Kleiderman, Ravitsky and Knoppers”, Beriain 2019
- “Transgenic Metarhizium Rapidly Kills Mosquitoes in a Malaria-Endemic Region of Burkina Faso”, Lovett et al 2019
- “Synthetic Glycolate Metabolism Pathways Stimulate Crop Growth and Productivity in the Field”, South & Cavanagh 2019
- “Principles of and Strategies for Germline Gene Therapy”, Wolf et al 2019
- “Cytosine Base Editor Generates Substantial Off-Target Single-Nucleotide Variants in Mouse Embryos”, Zuo et al 2019
- “Radical Technology Meets Radical Application: An Interview With George Church”, Davies & Church 2019
- “A DNA-Of-Things Storage Architecture to Create Materials With Embedded Memory”, Koch 2019
- “Human Germline Genome Editing”, Lea & Niakan 2019
- “Unbiased Detection of CRISPR Off-Targets in Vivo Using DISCOVER-Seq”, Wienert et al 2019
- “Reshuffling Yeast Chromosomes With CRISPR/Cas9”, Fleiss et al 2019
- “Analysis of Polygenic Score Usage and Performance across Diverse Human Populations”, Duncan et al 2018
- “Creating a Functional Single-Chromosome Yeast”, Shao et al 2018
- “Unleashing Meiotic Crossovers in Crops”, Mieulet et al 2018
- “In Vivo CRISPR Editing With No Detectable Genome-Wide Off-Target Mutations”, Akcakaya et al 2018
- “Gene Editing Restores Dystrophin Expression in a Canine Model of Duchenne Muscular Dystrophy”, Amoasii1 et al 2018
- “Accurate Classification of BRCA1 Variants With Saturation Genome Editing”, Findlay et al 2018
- “Genome-Edited Skin Epidermal Stem Cells Protect Mice from Cocaine-Seeking Behavior and Cocaine Overdose”, Li et al 2018
- “Reprogramming Human T Cell Function and Specificity With Non-Viral Genome Targeting”, Roth et al 2018
- “De Novo Domestication of Wild Tomato Using Genome Editing”, Zsögön et al 2018
- “Refining the Accuracy of Validated Target Identification through Coding Variant Fine-Mapping in Type 2 Diabetes”, Mahajan et al 2018
- “In Utero CRISPR-Mediated Therapeutic Editing of Metabolic Genes”, Rossidis et al 2018
- “Livestock 2.0—Genome Editing for Fitter, Healthier, and More Productive Farmed Animals”, Tait-Burkard et al 2018
- “Karyotype Engineering by Chromosome Fusion Leads to Reproductive Isolation in Yeast”, Luo et al 2018
- “Chinese Firm Clones Gene-Edited Dog in Bid to Treat Cardiovascular Disease”, Wang et al 2017
- “Polygenic Prediction of the Phenome, across Ancestry, in Emerging Adulthood”, Docherty et al 2017
- “Quantification of Frequency-Dependent Genetic Architectures and Action of Negative Selection in 25 UK Biobank Traits”, Schoech et al 2017
- “Genome-Wide Association Study Identifies 112 New Loci for Body Mass Index in the Japanese Population”, Akiyama et al 2017
- “Linkage Disequilibrium-Dependent Architecture of Human Complex Traits Shows Action of Negative Selection”, Gazal et al 2017
- “Inactivation of Porcine Endogenous Retrovirus in Pigs Using CRISPR-Cas9”, Niu et al 2017
- “Correction of a Pathogenic Gene Mutation in Human Embryos”, Ma et al 2017
- “Comparing Distributions of Polygenic Risk Scores of Type 2 Diabetes and Coronary Heart Disease within Different Populations”, Reisberg et al 2017
- “Nanoparticle Delivery of Cas9 Ribonucleoprotein and Donor DNA in Vivo Induces Homology-Directed DNA Repair”, Lee et al 2017
- “Implications of Human Genetic Variation in CRISPR-Based Therapeutic Genome Editing”, Scott & Zhang 2017
- “CRISPR/Cas9-Mediated Gene Editing in Human Zygotes Using Cas9 Protein”, Tang 2017
- “In Vivo Excision of HIV-1 Provirus by SaCas9 and Multiplex Single-Guide RNAs in Animal Models”, Yin et al 2017
- “Engineering Photosynthesis: Progress and Perspectives”, Orr et al 2017
- “Generation of Human Organs in Pigs via Interspecies Blastocyst Complementation”, Wu et al 2016
- “Total Biosynthesis of Opiates by Stepwise Fermentation Using Engineered Escherichia Coli”, Nakagawa et al 2016
- “Introducing Precise Genetic Modifications into Human 3PN Embryos by CRISPR/Cas-Mediated Genome Editing”, Kang et al 2016
- “High-Fidelity CRISPR–Cas9 Nucleases With No Detectable Genome-Wide Off-Target Effects”, Kleinstiver et al 2016
- “Schizophrenia Risk from Complex Variation of Complement Component 4”, Sekar et al 2016
- “Gene-Edited Pigs Are Protected from Porcine Reproductive and Respiratory Syndrome Virus”, Whitworth et al 2016
- “Therapeutic Genome Editing by Combined Viral and Non-Viral Delivery of CRISPR System Components in Vivo”, Yin et al 2016
- “In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa”, Bakondi et al 2016
- “Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage”, Komor et al 2016
- “From the Concept of Totipotency to Biofortified Cereals”, Potrykus 2015
- “CRISPR/Cas9-Mediated Gene Editing in Human Tripronuclear Zygotes”, Liang et al 2015
- “RNA-Guided Gene Drives Can Efficiently and Reversibly Bias Inheritance in Wild Yeast”, DiCarlo et al 2015
- “Proxy-Phenotype Method Identifies Common Genetic Variants Associated With Cognitive Performance”, Rietveld 2015
- “Update on the First Cloned Dog and Outlook for Canine Cloning”, Goo & ByeongChun 2015
- “Estimating the Mutation Load in Human Genomes”, Henn et al 2015
- “Genome Editing. The Mutagenic Chain Reaction: a Method for Converting Heterozygous to Homozygous Mutations”, Gantz & Bier 2015
- “Chromosome Transplantation As a Novel Approach for Correcting Complex Genomic Disorders”, Paulis et al 2015
- “One Hundred Years of Statistical Developments in Animal Breeding”, Gianola & Rosa 2014
- “Efficient Gene Knockout in Goats Using CRISPR/Cas9 System”, Ni et al 2014
- “The Deleterious Mutation Load Is Insensitive to Recent Population History”, Simons et al 2014
- “A Novel BHLHE41 Variant Is Associated With Short Sleep and Resistance to Sleep Deprivation in Humans”, Pellegrino et al 2014
- “Simultaneous Editing of Three Homoeoalleles in Hexaploid Bread Wheat Confers Heritable Resistance to Powdery Mildew”, Wang 2014
- “Molecular Genetic Evidence for Overlap between General Cognitive Ability and Risk for Schizophrenia: a Report from the Cognitive Genomics ConsorTium (COGENT)”, Lencz et al 2014
- “Genome Editing With Cas9 in Adult Mice Corrects a Disease Mutation and Phenotype”, Yin et al 2014
- “Sequence-Specific Antimicrobials Using Efficiently Delivered RNA-Guided Nucleases”, Citorik et al 2014
- “A Competitive Advantage by Neonatally Engrafted Human Glial Progenitors Yields Mice Whose Brains Are Chimeric for Human Glia”, Windrem et al 2014
- “Prevention of Muscular Dystrophy in Mice by CRISPR/Cas9-Mediated Editing of Germline DNA”, Long et al 2014
- “Generalization and Dilution of Association Results from European GWAS in Populations of Non-European Ancestry: The PAGE Study”, Carlson et al 2013
- “Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients”, Schwank et al 2013
- “RNA-Programmed Genome Editing in Human Cells”, Jinek et al 2013
- “Efficient Genome Editing in Zebrafish Using a CRISPR-Cas System”, Hwang et al 2013
- “RNA-Guided Human Genome Engineering via Cas9”, Mali et al 2013
- “Efficient Nonmeiotic Allele Introgression in Livestock Using Custom Endonucleases”, Tan 2013
- “Heritability of Performance Deficit Accumulation during Acute Sleep Deprivation in Twins”, Kuna et al 2012
- “A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity”, Jinek 2012
- “Conversion of Mouse Fibroblasts into Cardiomyocytes Using a Direct Reprogramming Strategy”, Efe et al 2011
- “The Transcriptional Repressor DEC2 Regulates Sleep Length in Mammals”, He et al 2009
- “Genetically Modified Streptococcus Mutans for the Prevention of Dental Caries”, Hillman 2002
- “Genetic Manipulation in Humans As a Matter of Rawlsian Justice”, Brown 2001
- “Young Receptors Make Smart Mice”, Bliss 1999
- “Genetic Enhancement of Learning and Memory in Mice”, Tang et al 1999
- “Cavalry in the Age of the Autarch”, Wolfe 1995
- “Book Review: Barriers to Bioweapons”
- “Some Thoughts on Education and Political Priorities, Cummings 2013”
- “A Sleep Diary and Questionnaire Study of Naturally Short Sleepers”
- “A Gene That Makes You Need Less Sleep?”
- Sort By Magic
- Wikipedia
- Miscellaneous
- Link Bibliography
See Also
Links
“Design of Highly Functional Genome Editors by Modeling the Universe of CRISPR-Cas Sequences”, Ruffolo et al 2024
Design of highly functional genome editors by modeling the universe of CRISPR-Cas sequences
“AAV1-HOTOF Gene Therapy for Autosomal Recessive Deafness 9: a Single-Arm Trial”, Lv et al 2024
AAV1-hOTOF gene therapy for autosomal recessive deafness 9: a single-arm trial
“Cryptography in the DNA of Living Cells Enabled by Multi-Site Base Editing”, Volf et al 2023
Cryptography in the DNA of living cells enabled by multi-site base editing
“GDNF Gene Therapy for Alcohol Use Disorder in Male Non-Human Primates”, Ford et al 2023b
GDNF gene therapy for alcohol use disorder in male non-human primates
“In Vivo Biosynthesis of N,N-Dimethyltryptamine, 5-MeO-N,N-Dimethyltryptamine, and Bufotenine in E. Coli”, Friedberg et al 2023
“Temperature-Dependent RNA Editing in Octopus Extensively Recodes the Neural Proteome”, Birk et al 2023
Temperature-dependent RNA editing in octopus extensively recodes the neural proteome
“National and Global Impacts of Genetically Modified Crops”, Hansen & Wingender 2023
“The First Crispr-Edited Salad Is Here: A Startup Used Gene Editing to Make Mustard Greens More Appetizing to Consumers. Next Up: Fruits”, Mullin 2023
“First Gene-Edited Calf With Reduced Susceptibility to a Major Viral Pathogen”, Workman et al 2023
First gene-edited calf with reduced susceptibility to a major viral pathogen
“Acceptance of Genetic Editing and of Whole Genome Sequencing of Human Embryos by Patients With Infertility Before and After the Onset of the COVID-19 Pandemic”, Neuhausser et al 2023
“CRISPR Technology: A Decade of Genome Editing Is Only the Beginning”, Wang & Doudna 2023
CRISPR technology: A decade of genome editing is only the beginning
“Gene Therapy Mediated Partial Reprogramming Extends Lifespan and Reverses Age-Related Changes in Aged Mice”, Macip et al 2023
“Gene-Edited Hens May End Cull of Billions of Chicks”, Ghosh 2022
“Base Editing: Revolutionary Therapy Clears Girl’s Incurable Cancer”, Gallagher 2022
Base editing: Revolutionary therapy clears girl’s incurable cancer
“MicroRNAs Are Deeply Linked to the Emergence of the Complex Octopus Brain”, Zolotarov et al 2022
MicroRNAs are deeply linked to the emergence of the complex octopus brain
“PASTE: Drag-And-Drop Genome Insertion of Large Sequences without Double-Strand DNA Cleavage Using CRISPR-Directed Integrases”, Yarnall et al 2022
“Large Scale Functional Screen Identifies Genetic Variants With Splicing Effects in Modern and Archaic Humans”, Rong et al 2022
“Prime Editing for Precise and Highly Versatile Genome Manipulation”, Chen & Liu 2022b
Prime editing for precise and highly versatile genome manipulation
“Role of Spike in the Pathogenic and Antigenic Behavior of SARS-CoV-2 BA.1 Omicron”, Chen et al 2022
Role of spike in the pathogenic and antigenic behavior of SARS-CoV-2 BA.1 Omicron
“New Self-Sexing Aedes Aegypti Strain Eliminates Barriers to Scalable and Sustainable Vector Control for Governments and Communities in Dengue-Prone Environments”, Spinner et al 2022
“RNA Recoding in Cephalopods Tailors Microtubule Motor Protein Function”, Rangan & Reck-Peterson 2022
RNA recoding in cephalopods tailors microtubule motor protein function
“A Sustainable Mouse Karyotype Created by Programmed Chromosome Fusion”, Wang et al 2022e
A sustainable mouse karyotype created by programmed chromosome fusion
“CRISPR-Cas9-Mediated Gene Editing of the BCL11A Enhancer for Pediatric Β0/β0 Transfusion-Dependent Β-Thalassemia”, Fu et al 2022
“A Versatile, High-Efficiency Platform for CRISPR-Based Gene Activation”, Heidersbach et al 2022
A versatile, high-efficiency platform for CRISPR-based gene activation
“Rubisco Function, Evolution, and Engineering”, Prywes et al 2022
“Generation of Genome-Edited Dogs by Somatic Cell Nuclear Transfer”, Kim et al 2022
Generation of genome-edited dogs by somatic cell nuclear transfer
“Therapeutic in Vivo Delivery of Gene Editing Agents”, Raguram et al 2022
“Retro-Cascorder: Recording Gene Expression Order in DNA by CRISPR Addition of Retron Barcodes”, Bhattarai-Kline et al 2022
Retro-Cascorder: Recording gene expression order in DNA by CRISPR addition of retron barcodes
“Genome-Edited Crops for Improved Food Security of Smallholder Farmers”, Pixley et al 2022
Genome-edited crops for improved food security of smallholder farmers
“The Creator of the CRISPR Babies Has Been Released from a Chinese Prison: He Jiankui Created the First Gene-Edited Children. The Price Was His Career. And His Freedom.”, Regalado 2022
“A CRISPR Kitty? Gene Editing Breathes New Life into the Hypoallergenic Cat”, Lin 2022
A CRISPR Kitty? Gene Editing Breathes New Life into the Hypoallergenic Cat
“Virgin Birth: A Genetic Basis for Facultative Parthenogenesis”, Braun et al 2022
Virgin Birth: A genetic basis for facultative parthenogenesis
“Viable Offspring Derived from Single Unfertilized Mammalian Oocytes”, Wei et al 2022b
Viable offspring derived from single unfertilized mammalian oocytes
“How to Protect the First ‘CRISPR Babies’ Prompts Ethical Debate: Fears of Excessive Interference Cloud Proposal for Protecting Children Whose Genomes Were Edited, As He Jiankui’s Release from Jail Looks Imminent”, Mallapaty 2022
“In Vivo Mitochondrial Base Editing via Adeno-Associated Viral Delivery to Mouse Post-Mitotic Tissue”, Silva-Pinheiro et al 2022
In vivo mitochondrial base editing via adeno-associated viral delivery to mouse post-mitotic tissue
“The Gene TaWOX5 Overcomes Genotype Dependency in Wheat Genetic Transformation”, Wang et al 2022
The gene TaWOX5 overcomes genotype dependency in wheat genetic transformation
“Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease”, Kanter et al 2021
Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease
“CRISPR-Cas9 Effectors Facilitate Generation of Single-Sex Litters and Sex-Specific Phenotypes”, Douglas et al 2021
CRISPR-Cas9 effectors facilitate generation of single-sex litters and sex-specific phenotypes
“Lipid Nanoparticles Incorporating a GalNAc Ligand Enable in Vivo Liver ANGPTL3 Editing in Wild-Type and Somatic LDLR Knockout Non-Human Primates”, Kasiewicz et al 2021
“A Temporally Resolved, Multiplex Molecular Recorder Based on Sequential Genome Editing”, Choi et al 2021
A temporally resolved, multiplex molecular recorder based on sequential genome editing
“Multiplex Genomic Recording of Enhancer and Signal Transduction Activity in Mammalian Cells”, Chen et al 2021
Multiplex genomic recording of enhancer and signal transduction activity in mammalian cells
“Drag-And-Drop Genome Insertion without DNA Cleavage With CRISPR-Directed Integrases”, Ioannidi et al 2021
Drag-and-drop genome insertion without DNA cleavage with CRISPR-directed integrases
“PRIME-Del: Precise Genomic Deletions Using Paired Prime Editing”, Choi et al 2021
PRIME-Del: Precise genomic deletions using paired prime editing
“PEDAR: Deletion and Replacement of Long Genomic Sequences Using Prime Editing”, Jiang et al 2021
PEDAR: Deletion and replacement of long genomic sequences using prime editing
“In a First, Surgeons Attached a Pig Kidney to a Human, and It Worked: A Kidney Grown in a Genetically Altered Pig Functions Normally, Scientists Reported. The Procedure May Open the Door to a Renewable Source of Desperately Needed Organs”, Rabin 2021
“The Widespread IS200/605 Transposon Family Encodes Diverse Programmable RNA-Guided Endonucleases”, Altae-Tran et al 2021
The widespread IS200/605 transposon family encodes diverse programmable RNA-guided endonucleases
“Mammalian Retrovirus-Like Protein PEG10 Packages Its Own MRNA and Can Be Pseudotyped for MRNA Delivery”, Segel et al 2021
“Gene-Drive Suppression of Mosquito Populations in Large Cages As a Bridge between Lab and Field”, Hammond et al 2021
Gene-drive suppression of mosquito populations in large cages as a bridge between lab and field
“RNA Demethylation Increases the Yield and Biomass of Rice and Potato Plants in Field Trials”, Yu et al 2021b
RNA demethylation increases the yield and biomass of rice and potato plants in field trials
“Surrogate Broodstock to Enhance Biotechnology Research and Applications in Aquaculture”, Jin et al 2021
Surrogate broodstock to enhance biotechnology research and applications in aquaculture
“CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis”, Gillmore et al 2021
CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis
“Base Editing of Haematopoietic Stem Cells Rescues Sickle Cell Disease in Mice”, Newby et al 2021
Base editing of haematopoietic stem cells rescues sickle cell disease in mice
“Scientists Drove Mice to Bond by Zapping Their Brains With Light: The Study, a Tour De Force in Bioengineering, Comes After Two Decades of Research on Brain-To-Brain Synchrony in People”, Hughes 2021
“Partial Recovery of Visual Function in a Blind Patient After Optogenetic Therapy”, Sahel et al 2021
Partial recovery of visual function in a blind patient after optogenetic therapy
“With Engineered Proteins, Scientists Use Optogenetics for the First Time to Help a Blind Patient See Again”, Molteni 2021
“In Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates”, Musunuru et al 2021
In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates
“High-Throughput Functional Variant Screens via in Vivo Production of Single-Stranded DNA”, Schubert et al 2021
High-throughput functional variant screens via in vivo production of single-stranded DNA
“First Genetically Modified Mosquitoes Released in the United States: Biotech Firm Oxitec Launches Controversial Field Test of Its Insects in Florida After Years of Push-Back from Residents and Regulatory Complications”, Waltz 2021
“Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing”, Nuñez et al 2021
Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing
“China Officially Bans CRISPR Babies, Human Clones and Animal-Human Hybrids”, News 2021
China officially bans CRISPR babies, human clones and animal-human hybrids
“Reintroduction of the Archaic Variant of NOVA1 in Cortical Organoids Alters Neurodevelopment”, Trujillo et al 2021
Reintroduction of the archaic variant of NOVA1 in cortical organoids alters neurodevelopment
“Reactivation of the Pluripotency Program Precedes Formation of the Cranial Neural Crest”, Zalc et al 2021
Reactivation of the pluripotency program precedes formation of the cranial neural crest
“In Vivo Base Editing Rescues Hutchinson-Gilford Progeria Syndrome in Mice”, Koblan et al 2021
In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice
“Expression of Functional Plant Sweet Protein Thaumatin II in the Milk of Transgenic Mice”, Lu et al 2021
Expression of functional plant sweet protein thaumatin II in the milk of transgenic mice
“Xenogeneic Stem Cell Transplantation: Research Progress and Clinical Prospects”, Jiang et al 2021
Xenogeneic stem cell transplantation: Research progress and clinical prospects
“Human-Animal Interspecies Chimerism via Blastocyst Complementation: Advances, Challenges and Perspectives: a Narrative Review”, Li & Huang 2021
“Reconstitution of the Oocyte Transcriptional Network With Transcription Factors”, Hamazaki et al 2020
Reconstitution of the oocyte transcriptional network with transcription factors
“Biotechnology Research Viewed With Caution Globally, but Most Support Gene Editing for Babies To Treat Disease: Majorities across Global Publics Accept Evolution; Religion Factors Prominently in Belief”, Funk et al 2020
“Reprogramming to Recover Youthful Epigenetic Information and Restore Vision”, Lu et al 2020
Reprogramming to recover youthful epigenetic information and restore vision
“Inclusion of Variants Discovered from Diverse Populations Improves Polygenic Risk Score Transferability”, Cavazos & Witte 2020
“Recency Negativity: Newer Food Crops Are Evaluated Less Favorably”, Inbar et al 2020
Recency negativity: Newer food crops are evaluated less favorably
“CRISPR-Enhanced Human Adipocyte ‘Browning’ As Cell Therapy for Metabolic Disease”, Tsagkaraki et al 2020
CRISPR-enhanced human adipocyte ‘browning’ as cell therapy for metabolic disease
“Press Release: The Nobel Prize in Chemistry 2020”, Sciences 2020
“Heritable Human Genome Editing”, Report 2020
“Human Embryo Gene Editing Gets a Road Map—Not a Green Light: After the 2018 ‘Crispr Baby’ Scandal, a Global Commission Assessed the Technology and Set Strict Criteria for Moving It toward Clinical Trials”, Molteni 2020
“CRISPR-Engineered Human Brown-Like Adipocytes Prevent Diet-Induced Obesity and Ameliorate Metabolic Syndrome in Mice”, Wang et al 2020d
“An Antiviral Self-Replicating Molecular Heterotroph”, Shapiro et al 2020
“Human-Specific ARHGAP11B Increases Size and Folding of Primate Neocortex in the Fetal Marmoset”, Heide et al 2020
Human-specific ARHGAP11B increases size and folding of primate neocortex in the fetal marmoset
“Cas9 Cuts and Consequences; Detecting, Predicting, and Mitigating CRISPR/Cas9 On-Target and Off-Target Damage [Techniques for Detecting, Predicting, and Mitigating the On-Target and Off-Target Effects of Cas9 Editing]”, Newman et al 2020
“A Bacterial Cytidine Deaminase Toxin Enables CRISPR-Free Mitochondrial Base Editing”, Mok et al 2020
A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing
“A Year In, 1st Patient To Get Gene Editing For Sickle Cell Disease Is Thriving”, Stein 2020
A Year In, 1st Patient To Get Gene Editing For Sickle Cell Disease Is Thriving
“Three People With Inherited Diseases Successfully Treated With CRISPR”, Page 2020
Three people with inherited diseases successfully treated with CRISPR
“CRISPR-Cas9-Mediated Induction of Heritable Chromosomal Translocations in Arabidopsis”, Beying et al 2020
CRISPR-Cas9-mediated induction of heritable chromosomal translocations in Arabidopsis
“Reversal of Aging via in Vivo Epigenetic Reprogramming”, Lu 2020b
“Generation of Human Endothelium in Pig Embryos Deficient in ETV2”, Das et al 2020
Generation of human endothelium in pig embryos deficient in ETV2
“The Promise and Challenge of Therapeutic Genome Editing”, Doudna 2020
The promise and challenge of therapeutic genome editing:
View PDF:
“Metabolic Engineering of Saccharomyces Cerevisiae for the de Novo Production of Psilocybin and Related Tryptamine Derivatives”, Milne et al 2020
“Chinese Scientist Who Genetically Edited Babies Gets 3 Years in Prison: He Jiankui’s Work Was Also Carried out on a Third Infant, according to China’s State Media, in a New Disclosure That Is Likely to Add to the Global Uproar over Such Experiments.”, Wee 2019
“Extensive Mammalian Germline Genome Engineering”, Yue et al 2019
“Eyeing Organs for Human Transplants, Companies Unveil the Most Extensively Gene-Edited Pigs Yet”, Servick 2019
Eyeing organs for human transplants, companies unveil the most extensively gene-edited pigs yet
“A Single Combination Gene Therapy Treats Multiple Age-Related Diseases”, Davidsohn et al 2019
A single combination gene therapy treats multiple age-related diseases
“Search-And-Replace Genome Editing without Double-Strand Breaks or Donor DNA”, Anzalone et al 2019
Search-and-replace genome editing without double-strand breaks or donor DNA
“New ‘Prime’ Genome Editor Could Surpass CRISPR”, Cohen 2019
“A New Crispr Technique Could Fix Almost All Genetic Diseases: A Less Error-Prone DNA Editing Method Could Correct Many More Harmful Mutations Than Was Previously Possible”, Molteni 2019
“Technological Challenges and Milestones for Writing Genomes: Synthetic Genomics Requires Improved Technologies”, Ostrov et al 2019
“CRISPR-Edited Stem Cells in a Patient With HIV and Acute Lymphocytic Leukemia”, Xu et al 2019
CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia
“The Promise and Price of Cellular Therapies: New ‘Living Drugs’—Made from a Patient’s Own Cells—Can Cure Once Incurable Cancers. But Can We Afford Them?”, Mukherjee 2019
“Russian Biologist Plans More CRISPR-Edited Babies: The Proposal Follows a Chinese Scientist Who Claimed to Have Created Twins from Edited Embryos Last Year”, Cyranoski 2019
“Exclusive: 5 Couples Lined up for CRISPR Babies to Avoid Deafness”, Page 2019
Exclusive: 5 couples lined up for CRISPR babies to avoid deafness
“Amid Animal Cruelty Debate, 80% of South Korea’s Sniffer Dogs Are Cloned”, Tribune 2019
Amid animal cruelty debate, 80% of South Korea’s sniffer dogs are cloned
“When Genome Editing Goes Off-Target: Detecting Unintended Mutations Could Improve DNA-Editing Strategies”, Kempton & Qi 2019
“Enabling Large-Scale Genome Editing by Reducing DNA Nicking”, Smith et al 2019
“Super-Mendelian Inheritance Mediated by CRISPR-Cas9 in the Female Mouse Germline”, Grunwald et al 2019
Super-Mendelian inheritance mediated by CRISPR-Cas9 in the female mouse germline
“The Transferability of Lipid-Associated Loci across African, Asian and European Cohorts”, Telkar et al 2019
The transferability of lipid-associated loci across African, Asian and European cohorts
“Improving Crop Yield: Synthetic Photorespiration Bypass Increases Crop Yield”, Eisenhut & Weber 2019
Improving crop yield: Synthetic photorespiration bypass increases crop yield
“Molecular Digital Data Storage Using DNA”, Ceze et al 2019
Molecular digital data storage using DNA:
View PDF:
“After the Storm—A Responsible Path for Genome Editing”, Daley et al 2019
After the Storm—A Responsible Path for Genome Editing:
View PDF:
“Is the ‘SErious’ Factor in Germline Modification Really Relevant? A Response to Kleiderman, Ravitsky and Knoppers”, Beriain 2019
“Transgenic Metarhizium Rapidly Kills Mosquitoes in a Malaria-Endemic Region of Burkina Faso”, Lovett et al 2019
Transgenic Metarhizium rapidly kills mosquitoes in a malaria-endemic region of Burkina Faso
“Synthetic Glycolate Metabolism Pathways Stimulate Crop Growth and Productivity in the Field”, South & Cavanagh 2019
Synthetic glycolate metabolism pathways stimulate crop growth and productivity in the field
“Principles of and Strategies for Germline Gene Therapy”, Wolf et al 2019
Principles of and strategies for germline gene therapy:
View PDF:
“Cytosine Base Editor Generates Substantial Off-Target Single-Nucleotide Variants in Mouse Embryos”, Zuo et al 2019
Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos
“Radical Technology Meets Radical Application: An Interview With George Church”, Davies & Church 2019
Radical Technology Meets Radical Application: An Interview with George Church:
View PDF:
“A DNA-Of-Things Storage Architecture to Create Materials With Embedded Memory”, Koch 2019
A DNA-of-things storage architecture to create materials with embedded memory:
View PDF:
“Human Germline Genome Editing”, Lea & Niakan 2019
Human germline genome editing:
View PDF:
“Unbiased Detection of CRISPR Off-Targets in Vivo Using DISCOVER-Seq”, Wienert et al 2019
Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq
“Reshuffling Yeast Chromosomes With CRISPR/Cas9”, Fleiss et al 2019
“Analysis of Polygenic Score Usage and Performance across Diverse Human Populations”, Duncan et al 2018
Analysis of Polygenic Score Usage and Performance across Diverse Human Populations
“Creating a Functional Single-Chromosome Yeast”, Shao et al 2018
“Unleashing Meiotic Crossovers in Crops”, Mieulet et al 2018
“In Vivo CRISPR Editing With No Detectable Genome-Wide Off-Target Mutations”, Akcakaya et al 2018
In vivo CRISPR editing with no detectable genome-wide off-target mutations:
View PDF:
“Gene Editing Restores Dystrophin Expression in a Canine Model of Duchenne Muscular Dystrophy”, Amoasii1 et al 2018
Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy
“Accurate Classification of BRCA1 Variants With Saturation Genome Editing”, Findlay et al 2018
Accurate classification of BRCA1 variants with saturation genome editing:
View PDF:
“Genome-Edited Skin Epidermal Stem Cells Protect Mice from Cocaine-Seeking Behavior and Cocaine Overdose”, Li et al 2018
View PDF:
“Reprogramming Human T Cell Function and Specificity With Non-Viral Genome Targeting”, Roth et al 2018
Reprogramming human T cell function and specificity with non-viral genome targeting:
View PDF:
“De Novo Domestication of Wild Tomato Using Genome Editing”, Zsögön et al 2018
De novo domestication of wild tomato using genome editing:
View PDF:
“Refining the Accuracy of Validated Target Identification through Coding Variant Fine-Mapping in Type 2 Diabetes”, Mahajan et al 2018
“In Utero CRISPR-Mediated Therapeutic Editing of Metabolic Genes”, Rossidis et al 2018
In utero CRISPR-mediated therapeutic editing of metabolic genes
“Livestock 2.0—Genome Editing for Fitter, Healthier, and More Productive Farmed Animals”, Tait-Burkard et al 2018
Livestock 2.0—genome editing for fitter, healthier, and more productive farmed animals
“Karyotype Engineering by Chromosome Fusion Leads to Reproductive Isolation in Yeast”, Luo et al 2018
Karyotype engineering by chromosome fusion leads to reproductive isolation in yeast
“Chinese Firm Clones Gene-Edited Dog in Bid to Treat Cardiovascular Disease”, Wang et al 2017
Chinese firm clones gene-edited dog in bid to treat cardiovascular disease
“Polygenic Prediction of the Phenome, across Ancestry, in Emerging Adulthood”, Docherty et al 2017
Polygenic prediction of the phenome, across ancestry, in emerging adulthood
“Quantification of Frequency-Dependent Genetic Architectures and Action of Negative Selection in 25 UK Biobank Traits”, Schoech et al 2017
“Genome-Wide Association Study Identifies 112 New Loci for Body Mass Index in the Japanese Population”, Akiyama et al 2017
Genome-wide association study identifies 112 new loci for body mass index in the Japanese population
“Linkage Disequilibrium-Dependent Architecture of Human Complex Traits Shows Action of Negative Selection”, Gazal et al 2017
“Inactivation of Porcine Endogenous Retrovirus in Pigs Using CRISPR-Cas9”, Niu et al 2017
Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9
“Correction of a Pathogenic Gene Mutation in Human Embryos”, Ma et al 2017
“Comparing Distributions of Polygenic Risk Scores of Type 2 Diabetes and Coronary Heart Disease within Different Populations”, Reisberg et al 2017
“Nanoparticle Delivery of Cas9 Ribonucleoprotein and Donor DNA in Vivo Induces Homology-Directed DNA Repair”, Lee et al 2017
View PDF:
“Implications of Human Genetic Variation in CRISPR-Based Therapeutic Genome Editing”, Scott & Zhang 2017
Implications of human genetic variation in CRISPR-based therapeutic genome editing:
View PDF:
“CRISPR/Cas9-Mediated Gene Editing in Human Zygotes Using Cas9 Protein”, Tang 2017
CRISPR/Cas9-mediated gene editing in human zygotes using Cas9 protein:
View PDF:
“In Vivo Excision of HIV-1 Provirus by SaCas9 and Multiplex Single-Guide RNAs in Animal Models”, Yin et al 2017
In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models
“Engineering Photosynthesis: Progress and Perspectives”, Orr et al 2017
“Generation of Human Organs in Pigs via Interspecies Blastocyst Complementation”, Wu et al 2016
Generation of human organs in pigs via interspecies blastocyst complementation
“Total Biosynthesis of Opiates by Stepwise Fermentation Using Engineered Escherichia Coli”, Nakagawa et al 2016
Total biosynthesis of opiates by stepwise fermentation using engineered Escherichia coli
“Introducing Precise Genetic Modifications into Human 3PN Embryos by CRISPR/Cas-Mediated Genome Editing”, Kang et al 2016
View PDF:
“High-Fidelity CRISPR–Cas9 Nucleases With No Detectable Genome-Wide Off-Target Effects”, Kleinstiver et al 2016
High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects:
“Schizophrenia Risk from Complex Variation of Complement Component 4”, Sekar et al 2016
Schizophrenia risk from complex variation of complement component 4
“Gene-Edited Pigs Are Protected from Porcine Reproductive and Respiratory Syndrome Virus”, Whitworth et al 2016
Gene-edited pigs are protected from porcine reproductive and respiratory syndrome virus
“Therapeutic Genome Editing by Combined Viral and Non-Viral Delivery of CRISPR System Components in Vivo”, Yin et al 2016
“In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa”, Bakondi et al 2016
“Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage”, Komor et al 2016
Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage
“From the Concept of Totipotency to Biofortified Cereals”, Potrykus 2015
“CRISPR/Cas9-Mediated Gene Editing in Human Tripronuclear Zygotes”, Liang et al 2015
CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes
“RNA-Guided Gene Drives Can Efficiently and Reversibly Bias Inheritance in Wild Yeast”, DiCarlo et al 2015
RNA-guided gene drives can efficiently and reversibly bias inheritance in wild yeast
“Proxy-Phenotype Method Identifies Common Genetic Variants Associated With Cognitive Performance”, Rietveld 2015
Proxy-Phenotype Method Identifies Common Genetic Variants Associated with Cognitive Performance
“Update on the First Cloned Dog and Outlook for Canine Cloning”, Goo & ByeongChun 2015
Update on the First Cloned Dog and Outlook for Canine Cloning:
View PDF:
“Estimating the Mutation Load in Human Genomes”, Henn et al 2015
“Genome Editing. The Mutagenic Chain Reaction: a Method for Converting Heterozygous to Homozygous Mutations”, Gantz & Bier 2015
“Chromosome Transplantation As a Novel Approach for Correcting Complex Genomic Disorders”, Paulis et al 2015
Chromosome transplantation as a novel approach for correcting complex genomic disorders
“One Hundred Years of Statistical Developments in Animal Breeding”, Gianola & Rosa 2014
One Hundred Years of Statistical Developments in Animal Breeding
“Efficient Gene Knockout in Goats Using CRISPR/Cas9 System”, Ni et al 2014
“The Deleterious Mutation Load Is Insensitive to Recent Population History”, Simons et al 2014
The deleterious mutation load is insensitive to recent population history
“A Novel BHLHE41 Variant Is Associated With Short Sleep and Resistance to Sleep Deprivation in Humans”, Pellegrino et al 2014
A novel BHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans
“Simultaneous Editing of Three Homoeoalleles in Hexaploid Bread Wheat Confers Heritable Resistance to Powdery Mildew”, Wang 2014
“Molecular Genetic Evidence for Overlap between General Cognitive Ability and Risk for Schizophrenia: a Report from the Cognitive Genomics ConsorTium (COGENT)”, Lencz et al 2014
“Genome Editing With Cas9 in Adult Mice Corrects a Disease Mutation and Phenotype”, Yin et al 2014
Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype
“Sequence-Specific Antimicrobials Using Efficiently Delivered RNA-Guided Nucleases”, Citorik et al 2014
Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases
“A Competitive Advantage by Neonatally Engrafted Human Glial Progenitors Yields Mice Whose Brains Are Chimeric for Human Glia”, Windrem et al 2014
“Prevention of Muscular Dystrophy in Mice by CRISPR/Cas9-Mediated Editing of Germline DNA”, Long et al 2014
Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA
“Generalization and Dilution of Association Results from European GWAS in Populations of Non-European Ancestry: The PAGE Study”, Carlson et al 2013
“Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients”, Schwank et al 2013
“RNA-Programmed Genome Editing in Human Cells”, Jinek et al 2013
“Efficient Genome Editing in Zebrafish Using a CRISPR-Cas System”, Hwang et al 2013
Efficient genome editing in zebrafish using a CRISPR-Cas system
“RNA-Guided Human Genome Engineering via Cas9”, Mali et al 2013
“Efficient Nonmeiotic Allele Introgression in Livestock Using Custom Endonucleases”, Tan 2013
Efficient nonmeiotic allele introgression in livestock using custom endonucleases
“Heritability of Performance Deficit Accumulation during Acute Sleep Deprivation in Twins”, Kuna et al 2012
Heritability of performance deficit accumulation during acute sleep deprivation in twins
“A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity”, Jinek 2012
A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity
“Conversion of Mouse Fibroblasts into Cardiomyocytes Using a Direct Reprogramming Strategy”, Efe et al 2011
Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy
“The Transcriptional Repressor DEC2 Regulates Sleep Length in Mammals”, He et al 2009
The transcriptional repressor DEC2 regulates sleep length in mammals
“Genetically Modified Streptococcus Mutans for the Prevention of Dental Caries”, Hillman 2002
Genetically modified Streptococcus mutans for the prevention of dental caries
“Genetic Manipulation in Humans As a Matter of Rawlsian Justice”, Brown 2001
Genetic Manipulation in Humans as a Matter of Rawlsian Justice
“Young Receptors Make Smart Mice”, Bliss 1999
“Genetic Enhancement of Learning and Memory in Mice”, Tang et al 1999
“Cavalry in the Age of the Autarch”, Wolfe 1995
“Book Review: Barriers to Bioweapons”
“Some Thoughts on Education and Political Priorities, Cummings 2013”
Some thoughts on education and political priorities, Cummings 2013
“A Sleep Diary and Questionnaire Study of Naturally Short Sleepers”
A sleep diary and questionnaire study of naturally short sleepers
“A Gene That Makes You Need Less Sleep?”
Sort By Magic
Annotations sorted by machine learning into inferred 'tags'. This provides an alternative way to browse: instead of by date order, one can browse in topic order. The 'sorted' list has been automatically clustered into multiple sections & auto-labeled for easier browsing.
Beginning with the newest annotation, it uses the embedding of each annotation to attempt to create a list of nearest-neighbor annotations, creating a progression of topics. For more details, see the link.
crispr
genomic-variation
gene-editing
Wikipedia
Miscellaneous
-
/doc/genetics/editing/2022-volcaniinstitute-goldahengeneeditingtoeliminatemalechicks.png: -
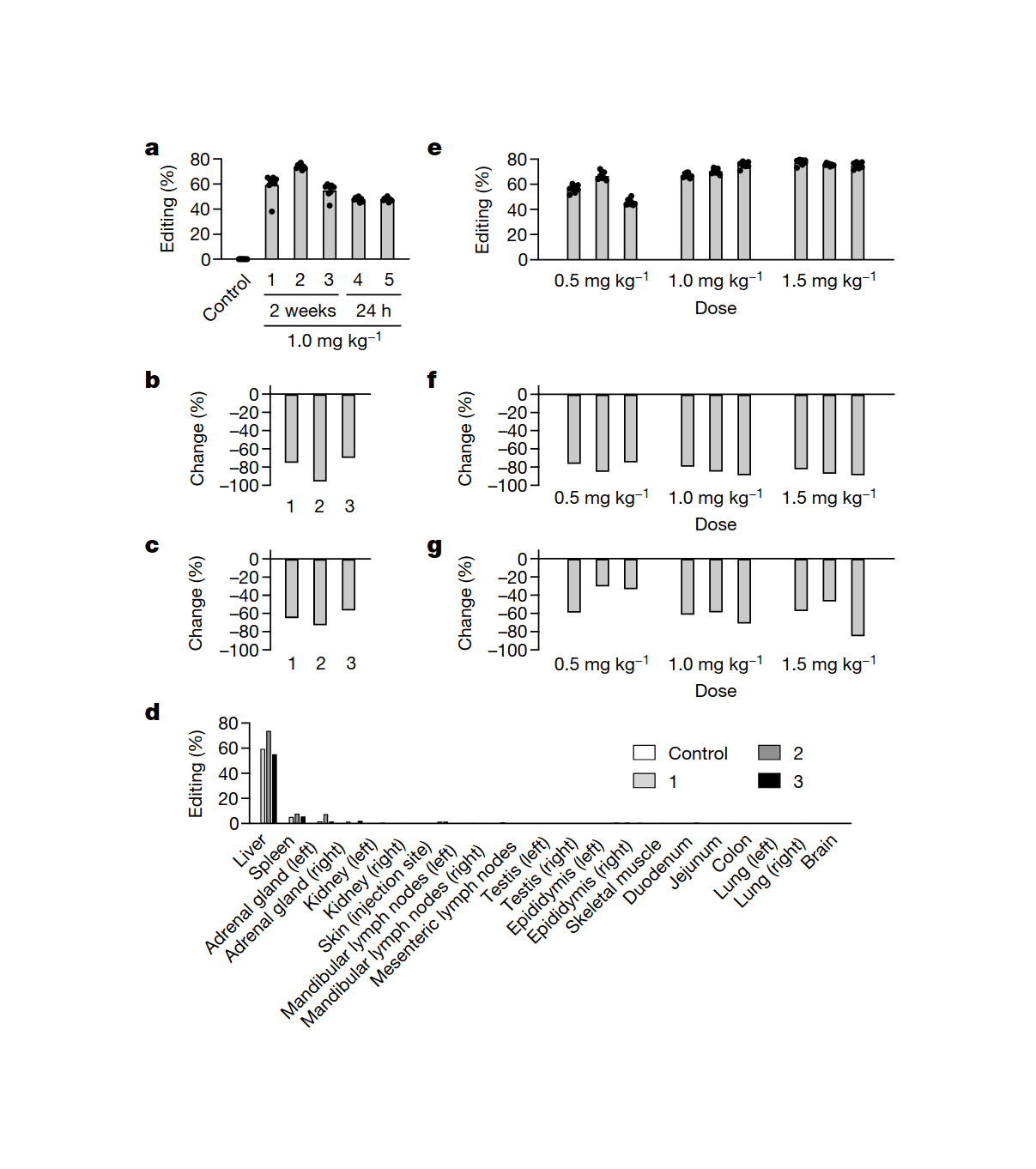
/doc/genetics/editing/2021-musunuru-figure2-shorttermeffectsofcrisprediting.png: -
/doc/genetics/editing/2021-musunuru-figure3-longtermeffectsofcrisprediting.png: -
/doc/genetics/editing/2019-miraclemilly-62019cv00425-complaint.pdf -
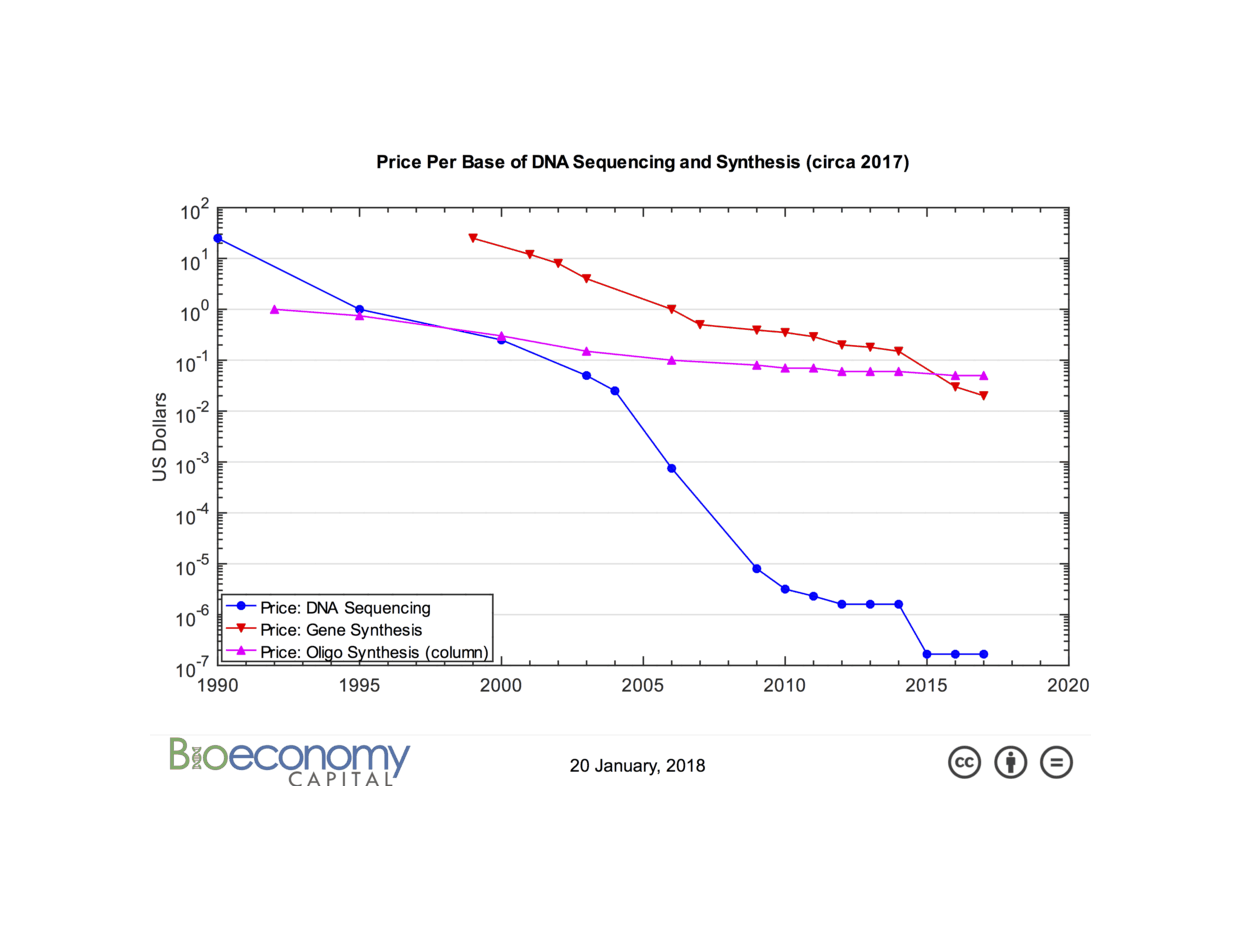
/doc/genetics/editing/2018-01-20-carlson-2017synthesiscostcurve.png: -
/doc/genetics/editing/2017-normile.pdf:View PDF:
-
/doc/genetics/editing/2017-scheufele.pdf:View PDF:
-
/doc/genetics/editing/2016-slaymaker.pdf:View PDF:
-
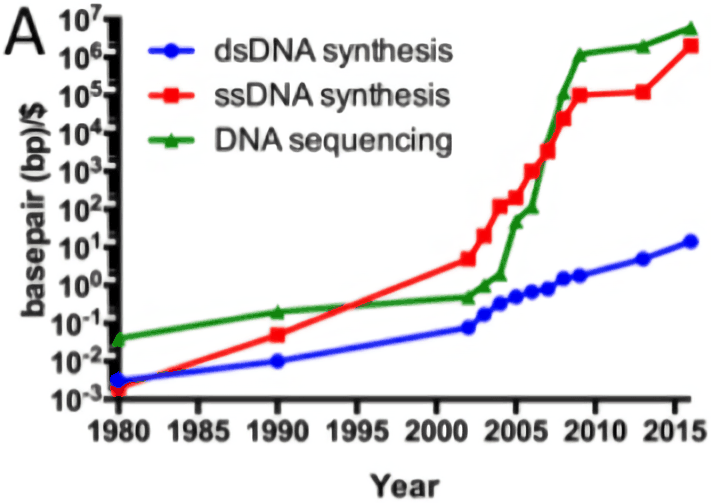
/doc/genetics/editing/2016-hgpwrite-carlson-synthesiscostcurve.png: -
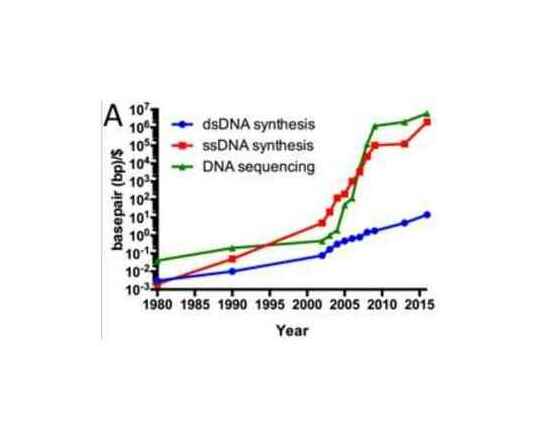
/doc/genetics/editing/2014-kosuri-genomesynthesis-costcurve.jpg: -
https://eryney.substack.com/p/what-are-the-bottlenecks-to-safe-935 -
https://forum.effectivealtruism.org/posts/nSwaDrHunt3ohh9Et/cause-area-short-sleeper-genes -
https://gsejournal.biomedcentral.com/articles/10.1186/s12711-016-0280-3 -
https://mainichi.jp/english/articles/20240329/p2a/00m/0sc/050000c -
https://maximumprogress.substack.com/p/grading-extropian-predictions -
https://www.lesswrong.com/posts/gjs3q83hA4giubaAw/will-no-one-rid-me-of-this-turbulent-pest: -
https://www.newyorker.com/magazine/2021/12/13/creating-a-better-leaf -
https://www.newyorker.com/magazine/2023/09/11/the-transformative-alarming-power-of-gene-editing -
https://www.newyorker.com/science/elements/will-plants-ever-fertilize-themselves -
https://www.nytimes.com/2024/01/23/health/deaf-gene-therapy.html -
https://www.nytimes.com/2024/03/21/health/pig-kidney-organ-transplant.html -
https://www.quantamagazine.org/how-genes-can-leap-from-snakes-to-frogs-20221027/ -
https://www.science.org/content/blog-post/more-crispr-human-subjects: -
https://www.technologyreview.com/2021/11/09/1039107/e-coli-maze-solving-biocomputer/: -
https://www.technologyreview.com/2023/10/27/1082551/gene-treatment-deaf-children-hearing-china/ -
https://www.wired.com/story/a-one-time-shot-for-type-2-diabetes-a-biotech-company-is-on-it/: -
https://www.wired.com/story/combined-heart-pump-pig-kidney-transplant-surgery/:View External Link:
https://www.wired.com/story/combined-heart-pump-pig-kidney-transplant-surgery/ -
https://www.wired.com/story/gene-editing-flies-to-fight-crop-damage/:View External Link:
https://www.wired.com/story/gene-editing-flies-to-fight-crop-damage/ -
https://www.wired.com/story/gene-therapy-in-the-womb-is-inching-closer-to-reality/: -
https://www.wired.com/story/gene-tweaked-jellyfish-neurology/ -
https://www.wired.com/story/the-secret-ingredient-in-your-craft-beer-gene-edited-yeast/
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Link Bibliography
-
2023-friedberg.pdf: “In Vivo Biosynthesis of N,N-Dimethyltryptamine, 5-MeO-N,N-Dimethyltryptamine, and Bufotenine in E. Coli”, -
https://www.cell.com/cell/fulltext/S0092-8674(23)00523-8: “Temperature-Dependent RNA Editing in Octopus Extensively Recodes the Neural Proteome”, -
2023-hansen.pdf: “National and Global Impacts of Genetically Modified Crops”, Casper Worm Hansen, Asger Mose Wingender -
https://www.wired.com/story/wired30-crispr-edited-salad-greens/: “The First Crispr-Edited Salad Is Here: A Startup Used Gene Editing to Make Mustard Greens More Appetizing to Consumers. Next Up: Fruits”, Emily Mullin -
https://www.biorxiv.org/content/10.1101/2023.01.04.522507.full: “Gene Therapy Mediated Partial Reprogramming Extends Lifespan and Reverses Age-Related Changes in Aged Mice”, Carolina Cano Macip, Rokib Hasan, Victoria Hoznek, Jihyun Kim, Louis E. Metzger, Saumil Sethna, Noah Davidsohn -
https://www.frontiersin.org/articles/10.3389/fbioe.2022.975786/full: “New Self-Sexing Aedes Aegypti Strain Eliminates Barriers to Scalable and Sustainable Vector Control for Governments and Communities in Dengue-Prone Environments”, -
2022-wang-5.pdf: “A Sustainable Mouse Karyotype Created by Programmed Chromosome Fusion”, -
https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-022-00749-3: “Generation of Genome-Edited Dogs by Somatic Cell Nuclear Transfer”, Dong-Ern Kim, Ji-Hye Lee, Kuk-Bin Ji, Kang-Sun Park, Tae-Young Kil, Okjae Koo, Min-Kyu Kim -
2022-pixley.pdf: “Genome-Edited Crops for Improved Food Security of Smallholder Farmers”, -
2022-wang.pdf: “The Gene TaWOX5 Overcomes Genotype Dependency in Wheat Genetic Transformation”, -
2019-kempton.pdf: “When Genome Editing Goes Off-Target: Detecting Unintended Mutations Could Improve DNA-Editing Strategies”, Hannah R. Kempton, Lei S. Qi -
2019-zuo.pdf: “Cytosine Base Editor Generates Substantial Off-Target Single-Nucleotide Variants in Mouse Embryos”,