- See Also
-
Links
- “Illusory Generalizability of Clinical Prediction Models”
- “Cat Ownership and Schizophrenia-Related Disorders and Psychotic-Like Experiences: A Systematic Review and Meta-Analysis: Supplement”, McGrath et al 2023
- “Cat Ownership and Schizophrenia-Related Disorders and Psychotic-Like Experiences: A Systematic Review and Meta-Analysis”, McGrath et al 2023
- “Rare Coding Variants in Schizophrenia-Associated Genes Affect Generalised Cognition in the UK Biobank”, Fenner et al 2023
- “Antipsychotic Drug Efficacy Correlates With the Modulation of D1 rather than D2 Receptor-Expressing Striatal Projection Neurons”, Yun et al 2023
- “Long-Acting Injectable Second-Generation Antipsychotics vs Placebo and Their Oral Formulations in Acute Schizophrenia: A Systematic Review and Meta-Analysis of Randomized-Controlled-Trials”, Wang et al 2023b
- “Longitudinal Follow-Up of Subsequent Psychiatric Comorbidities among Children and Adolescents With Autism Spectrum Disorder”, Yeh et al 2023
- “Common and Rare Variant Associations With Latent Traits Underlying Depression, Bipolar Disorder, and Schizophrenia”, Dattani et al 2023
- “Relationship of Family Genetic Risk Score With Diagnostic Trajectory in a Swedish National Sample of Incident Cases of Major Depression, Bipolar Disorder, Other Non-Affective Psychosis, and Schizophrenia”, Kendler et al 2023
- “Schizophrenia-Associated Somatic Copy-Number Variants from 12,834 Cases Reveal Recurrent NRXN1 and ABCB11 Disruptions”, Maury et al 2023
- “What Is the Frequency and Nature of Visual Hallucinations in Non-Clinical Participants?”, Aynsworth et al 2022
- “Polygenic Influences Associated With Adolescent Cognitive Skills”, Mitchell et al 2022
- “Voice Patterns As Markers of Schizophrenia: Building a Cumulative Generalizable Approach via a Cross-Linguistic and Meta-Analysis Based Investigation”, Parola et al 2022
- “Polygenic Architecture of Rare Coding Variation across 400,000 Exomes”, Weiner et al 2022
- “Is an Elevated Family-Genetic Risk for Major Psychiatric Disorders Specific to Creative Occupations?”, Kendler et al 2022b
- “Schizophrenia Polygenic Risk Score and Long-Term Success in the Labour Market: A Cohort Study”, Viinikainen et al 2022
- “The Course of General Cognitive Ability in Individuals With Psychotic Disorders”, Jonas et al 2022
- “Magical Thinking in Individuals With High Polygenic Risk for Schizophrenia but No Non-Affective Psychoses—A General Population Study”, Saarinen et al 2022
- “Multi-Omics Analyses Cannot Identify True-Positive Novel Associations from Underpowered Genome-Wide Association Studies of Four Brain-Related Traits”, Baranger et al 2022
- “The Relationship of Major Diseases With Childlessness: a Sibling Matched Case-Control and Population Register Study in Finland and Sweden”, Liu et al 2022
- “Occurrence of Psychosis and Bipolar Disorder in Adults With Autism: A Systematic Review and Meta-Analysis”, Varcin et al 2022
- “Electroconvulsive Therapy”, Espinoza & Kellner 2022
- “Genome-Wide Analyses of ADHD Identify 27 Risk Loci, Refine the Genetic Architecture and Implicate Several Cognitive Domains”, Demontis et al 2022
- “Neurocognitive Profile of Adolescents With Early-Onset Schizophrenia and Their Unaffected Siblings”, Vyas et al 2022
- “Interaction Testing and Polygenic Risk Scoring to Estimate the Association of Common Genetic Variants With Treatment Resistance in Schizophrenia”, Pardiñas et al 2022
- “Rare Schizophrenia Risk Variant Burden Is Conserved in Diverse Human Populations”, Liu et al 2022
- “Association of Schizophrenia Spectrum Disorders and Violence Perpetration in Adults and Adolescents from 15 Countries: A Systematic Review and Meta-Analysis”, Whiting et al 2021
- “Role of Polygenic Risk Score in the Familial Transmission of Bipolar Disorder in Youth”, Birmaher et al 2021
- “High-Impact Rare Genetic Variants in Severe Schizophrenia”, Zoghbi et al 2021
- “Familial Clustering of Psychiatric Disorders and Low IQ”, Weiser et al 2021
- “Deletion of Loss-Of-Function-Intolerant Genes and Risk of 5 Psychiatric Disorders”, Wainberg et al 2021
- “Comparing Copy Number Variations in a Danish Case Cohort of Individuals With Psychiatric Disorders”, Sánchez et al 2021
- “Novel Disease Associations With Schizophrenia Genetic Risk Revealed in ~400,000 UK Biobank Participants”, Zhang et al 2021b
- “A Selection Pressure Landscape for 870 Human Polygenic Traits”, Song et al 2021
- “A General Framework for Identifying Rare Variant Combinations in Complex Disorders”, Pounraja & Girirajan 2021
- “Investigating Perceived Heritability of Mental Health Disorders and Attitudes toward Genetic Testing in the United States, United Kingdom, and Australia”, Morosoli et al 2021
- “Independent Contribution of Polygenic Risk for Schizophrenia and Cannabis Use in Predicting Psychotic-Like Experiences in Young Adulthood: Testing Gene × Environment Moderation and Mediation”, Elkrief et al 2021
- “Adolescent Cannabis Use and Adult Psychoticism: A Longitudinal Co-Twin Control Analysis Using Data from Two Cohorts”, Schaefer et al 2021
- “The Prevalence of Mental Disorders among Homeless People in High-Income Countries: An Updated Systematic Review and Meta-Regression Analysis”, Gutwinski et al 2021
- “Genome-Wide Association Meta-Analysis of Childhood and Adolescent Internalizing Symptoms”, Jami et al 2021
- “Treating Cognitive Impairment in Schizophrenia With GLP-1RAs: an Overview of Their Therapeutic Potential”, Flintoff et al 2021
- “Cognitive Behavior Therapy for Depression From an Evolutionary Perspective”, Hollon et al 2021
- “Domestic Mass Shooters: The Association With Unmedicated and Untreated Psychiatric Illness”, Glick et al 2021
- “Career Effects of Mental Health”, Biasi et al 2021 (page 2)
- “Utility of Polygenic Embryo Screening for Disease Depends on the Selection Strategy”, Lencz et al 2021
- “Leroy’s Elusive Little People: A Systematic Review on Lilliputian Hallucinations”, Blom 2021
- “No Causal Associations between Childhood Family Income and Subsequent Psychiatric Disorders, Substance Misuse and Violent Crime Arrests: a Nationwide Finnish Study of >650 000 Individuals and Their Siblings”, Sariaslan et al 2021
- “Ultra-Rare, Rare, and Common Genetic Variant Analysis Converge to Implicate Negative Selection and Neuronal Processes in the Aetiology of Schizophrenia”, Akingbuwa et al 2021
- “The Relationship between Cannabis and Schizophrenia: a Genetically Informed Perspective”, Johnson et al 2021
- “A Comparison of Ten Polygenic Score Methods for Psychiatric Disorders Applied across Multiple Cohorts”, Ni et al 2021
- “Polygenic Risk for Depression, Anxiety and Neuroticism Are Associated With the Severity and Rate of Change in Depressive Symptoms across Adolescence”, Kwong et al 2021
- “Ditching Candidate Gene Association Studies: Lessons from Psychiatric Genetics”, Duarte et al 2021
- “Polygenic Burden Has Broader Impact on Health, Cognition, and Socioeconomic Outcomes Than Most Rare and High-Risk Copy Number Variants”, Saarentaus et al 2021
- “From Genotype to Phenotype: Polygenic Prediction of Complex Human Traits”, Raben et al 2021
- “Antidepressant Response in Major Depressive Disorder: A Genome-Wide Association Study”, Pain et al 2020
- “Ten Years of Enhancing Neuro-Imaging Genetics through Meta-Analysis: An Overview from the ENIGMA Genetics Working Group”, Medland et al 2020
- “The Comorbidity of Schizophrenia Spectrum and Mood Disorders in Autism Spectrum Disorder”, Chien et al 2020
- “Use of Genetically Informed Methods to Clarify the Nature of the Association Between Cannabis Use and Risk for Schizophrenia”, Gillespie & Kendler 2020
- “Violence and Mental Disorders: a Structured Review of Associations by Individual Diagnoses, Risk Factors, and Risk Assessment”, Whiting et al 2020
- “Genetic Architecture of 11 Major Psychiatric Disorders at Biobehavioral, Functional Genomic, and Molecular Genetic Levels of Analysis”, Grotzinger et al 2020
- “Exome Sequencing Identifies Rare Coding Variants in 10 Genes Which Confer Substantial Risk for Schizophrenia”, Singh et al 2020
- “Common Variants Contribute to Intrinsic Human Brain Functional Networks”, Zhao et al 2020
- “Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia”, Consortium et al 2020
- “Novel Ultra-Rare Exonic Variants Identified in a Founder Population Implicate Cadherins in Schizophrenia”, Lencz et al 2020
- “A Case of Mirror Image Agnosia and Mirrored Self-Misidentification Syndrome in Schizophrenia without Dementia or Structural Abnormalities”, Rong et al 2020
- “The Polygenic Architecture of Schizophrenia—Rethinking Pathogenesis and Nosology”, Smeland et al 2020
- “Risk in Relatives, Heritability, SNP-Based Heritability, and Genetic Correlations in Psychiatric Disorders: A Review”, Baselmans et al 2020
- “Obscure and Unknown: Deliriants of the Edgewood Arsenal Human Experiments”, space_crustacean 2020
- “Conditional GWAS Analysis to Identify Disorder-Specific SNPs for Psychiatric Disorders”, Byrne et al 2020
- “Disentangling Selection on Genetically Correlated Polygenic Traits Using Whole-Genome Genealogies”, Stern et al 2020
- “Need to Account for Familial Confounding in Systematic Review and Meta-Analysis of Prenatal Tobacco Smoke Exposure and Schizophrenia”, Quinn et al 2020
- “Is Early Blindness Protective of Psychosis or Are We Turning a Blind Eye to the Lack of Statistical Power?”, Jefsen et al 2020
- “Genome-Wide Association Study of Creativity Reveals Genetic Overlap With Psychiatric Disorders, Risk Tolerance, and Risky Behaviors”, Li et al 2020c
- “Genetics of Schizophrenia in the South African Xhosa”, Gulsuner et al 2020
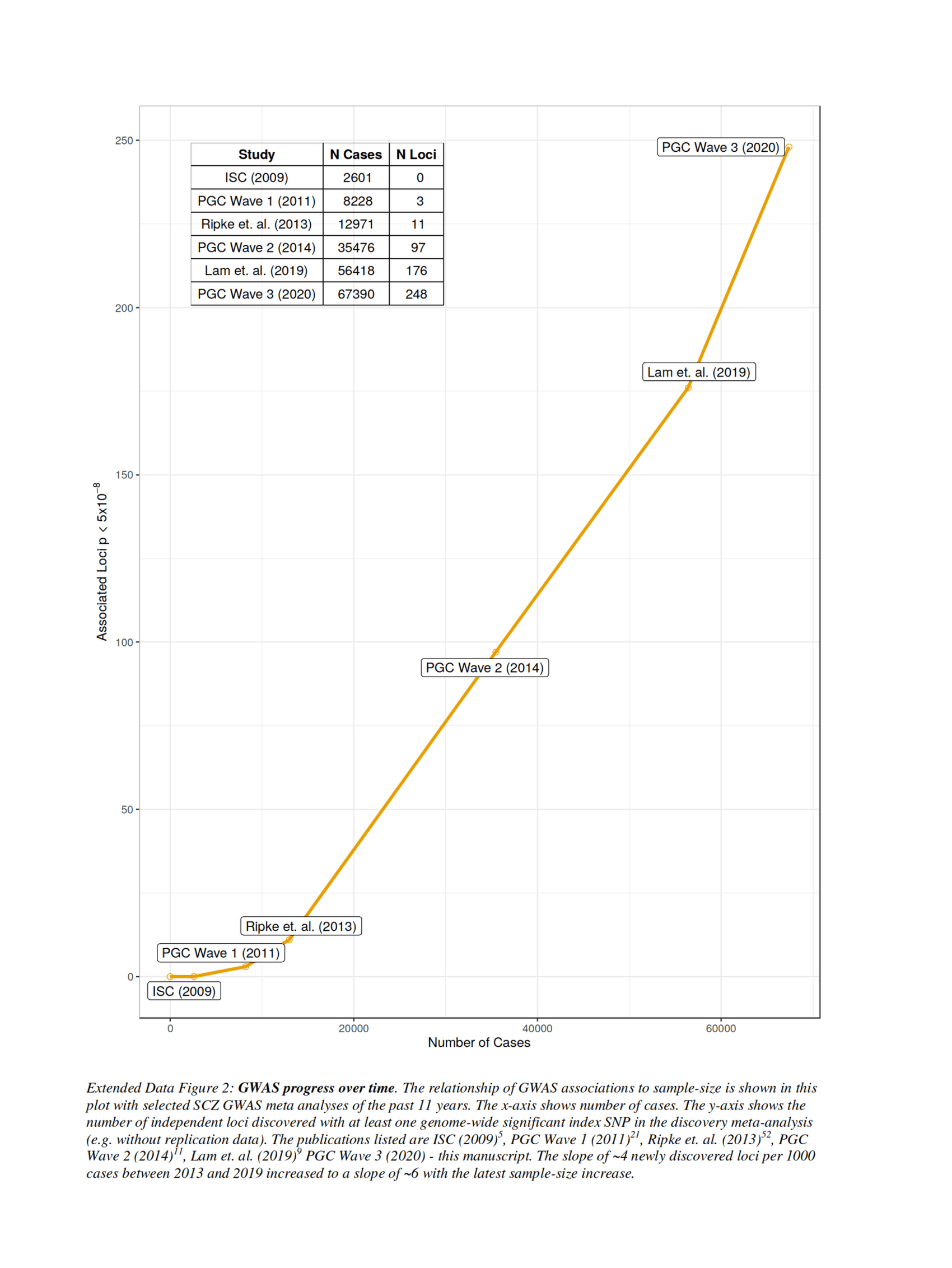
- “Extended Data Figure 2: GWAS Progress over Time. The Relationship of GWAS Associations to Sample-Size Is Shown in This Plot With Selected SCZ GWAS Meta-Analyses of the past 11 Years. The X-Axis Shows Number of Cases. The Y-Axis Shows the Number of Independent Loci Discovered With at Least One Genome-Wide Statistically-Significant Index SNP in the Discovery Meta-Analysis (eg. without Replication Data)...The Slope of ~4 Newly Discovered Loci per 1000 Cases 2013–2019 Increased to a Slope of ~6 With the Latest Sample-Size Increase.”
- “Risk of Subjection to Violence and Perpetration of Violence in Persons With Psychiatric Disorders in Sweden”, Sariaslan et al 2020
- “A Large-Scale Genome-Wide Association Study Meta-Analysis of Cannabis Use Disorder”, Johnson et al 2020
- “Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders”, Consortium 2019
- “Genome-Wide Association Studies in Schizophrenia: Recent Advances, Challenges and Future Perspective”, Dennison et al 2019
- “Genome-Wide Association Study Identifies 49 Common Genetic Variants Associated With Handedness”, Partida et al 2019
- “Risk of Non-Affective Psychotic Disorder or Bipolar Disorder in Autism Spectrum Disorder: a Longitudinal Register-Based Study in the Netherlands”, Schalbroeck et al 2019
- “Childhood Adoption and Mental Health in Adulthood: The Role of Gene-Environment Correlations and Interactions in the UK Biobank”, Lehto et al 2019
- “Unravelling the Genetic Basis of Schizophrenia and Bipolar Disorder With GWAS: A Systematic Review”, Prata et al 2019
- “Penetrance and Pleiotropy of Polygenic Risk Scores for Schizophrenia in 106,160 Patients across Four Healthcare Systems”, Zheutlin et al 2019
- “Evidence That the Association of Childhood Trauma With Psychosis and Related Psychopathology Is Not Explained by Gene-Environment Correlation: A Monozygotic Twin Differences Approach”, Lecei et al 2019
- “Psychiatric Comorbidity in Persons With High-Functioning Autism Spectrum Disorders: Findings from a Tertiary Care Neuropsychiatric Hospital”, Nahar et al 2019
- “Genome Wide Meta-Analysis Identifies Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders”, Consortium et al 2019
- “Phenotypic Annotation: Using Polygenic Scores to Translate Discoveries From Genome-Wide Association Studies From the Top Down”, Belsky & Harden 2019
- “Genome-Wide Analysis Reveals Extensive Genetic Overlap between Schizophrenia, Bipolar Disorder, and Intelligence”, Smeland et al 2019
- “The past and Future of Novel, Non-Dopamine-2 Receptor Therapeutics for Schizophrenia: A Critical and Comprehensive Review”, Girgis et al 2019
- “Common Polygenic Variations for Psychiatric Disorders and Cognition in Relation to Brain Morphology in the General Pediatric Population”, Alemany et al 2019
- “Schizophrenia and Bipolar Illness in the Relatives of University Scientists: An Epidemiological Report on the Creativity-Psychopathology Relationship”, Parnas et al 2019
- “Schizophrenia Risk and Reproductive Success: a Mendelian Randomization Study”, Lawn et al 2019
- “Schizophrenia Risk Conferred by Protein-Coding de Novo Mutations”, Howrigan et al 2018
- “Using Genetics to Examine a General Liability to Childhood Psychopathology”, Riglin et al 2018
- “The Genetic Relationship between Female Reproductive Traits and Six Psychiatric Disorders”, Ni et al 2018
- “Glucagon-Like Peptide-1 Receptor Agonists for Antipsychotic-Associated Cardio-Metabolic Risk Factors: A Systematic Review and Individual Participant Data Meta-Analysis”, Siskind et al 2018
- “Analysis of Polygenic Score Usage and Performance across Diverse Human Populations”, Duncan et al 2018
- “Genetics & the Geography of Health, Behavior, and Attainment”, Belsky et al 2018
- “An Evolutionary Compass for Elucidating Selection Mechanisms Shaping Complex Traits”, Uricchio et al 2018
- “Common Genetic Variants Contribute to Risk of Rare Severe Neurodevelopmental Disorders”, Niemi et al 2018
- “Evolutionary Perspectives on Genetic and Environmental Risk Factors for Psychiatric Disorders”, Keller 2018
- “Genetic Variants Associated With Subjective Well-Being, Depressive Symptoms, and Neuroticism Identified through Genome-Wide Analyses”, Okbay et al 2018
- “GWAS in 446,118 European Adults Identifies 78 Genetic Loci for Self-Reported Habitual Sleep Duration Supported by Accelerometer-Derived Estimates”, Dashti et al 2018
- “Genome-Wide Association Analyses of Chronotype in 697,828 Individuals Provides New Insights into Circadian Rhythms in Humans and Links to Disease”, Jones et al 2018
- “The Genetic Architecture of Schizophrenia, Bipolar Disorder, Obsessive-Compulsive Disorder and Autism Spectrum Disorder”, O’’Connell et al 2018
- “Improving Genetic Prediction by Leveraging Genetic Correlations among Human Diseases and Traits”, Maier et al 2018
- “Association Between Population Density and Genetic Risk for Schizophrenia”, Colodro-Conde et al 2018
- “Common Schizophrenia Alleles Are Enriched in Mutation-Intolerant Genes and in Regions under Strong Background Selection”, Pardiñas et al 2018
- “Genome-Wide Association Meta-Analysis in 269,867 Individuals Identifies New Genetic and Functional Links to Intelligence”, Savage et al 2018
- “Effects of Latent Toxoplasmosis on Olfactory Functions of Men and Women”, Flegr et al 2017
- “Polygenic Prediction of the Phenome, across Ancestry, in Emerging Adulthood”, Docherty et al 2017
- “Common Risk Variants Identified in Autism Spectrum Disorder”, Grove et al 2017
- “The Molecular Genetics of Participation in the Avon Longitudinal Study of Parents and Children”, Taylor et al 2017
- “Genome-Wide Association Study of Social Relationship Satisfaction: Loci and Correlations With Psychiatric Conditions”, Warrier et al 2017
- “Different Worlds”, Alexander 2017
- “Linkage Disequilibrium-Dependent Architecture of Human Complex Traits Shows Action of Negative Selection”, Gazal et al 2017
- “GWAS Meta-Analysis of Neuroticism (n = 449,484) Identifies Novel Genetic Loci and Pathways”, Nagel et al 2017
- “Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register”, Hilker et al 2017
- “Genome-Wide Analysis of Risk-Taking Behavior and Cross-Disorder Genetic Correlations in 116,255 Individuals from the UK Biobank Cohort”, Strawbridge et al 2017
- “Genome-Wide Association Study Identifies 30 Loci Associated With Bipolar Disorder”, Stahl et al 2017
- “Genomic Dissection of Bipolar Disorder and Schizophrenia including 28 Subphenotypes”, Ruderfer et al 2017
- “Genome-Wide Association Analyses Identify 44 Risk Variants and Refine the Genetic Architecture of Major Depressive Disorder”, Wray et al 2017
- “Quantifying the Impact of Rare and Ultra-Rare Coding Variation across the Phenotypic Spectrum”, Ganna et al 2017
- “Psychiatric Vulnerability in Adults With Intellectual Disability and Autism: A Literature Review”, Coli et al 2017
- “Genome-Wide Analysis of 113,968 Individuals in UK Biobank Identifies 4 Loci Associated With Mood Instability”, Ward et al 2017
- “No Cognitive-Enhancing Effect of GLP-1 Receptor Agonism in Antipsychotic-Treated, Obese Patients With Schizophrenia”, Ishøy et al 2017
- “Widespread Signatures of Positive Selection in Common Risk Alleles Associated to Autism Spectrum Disorder”, Polimanti & Gelernter 2017
- “Genetic and Environmental Contributions to the Association Between Cannabis Use and Psychotic-Like Experiences in Young Adult Twins”, Nesvåg et al 2017
- “The Structure and Measurement of Unusual Sensory Experiences in Different Modalities: The Multi-Modality Unusual Sensory Experiences Questionnaire (MUSEQ)”, Mitchell et al 2017
- “No Evidence That Schizophrenia Candidate Genes Are More Associated With Schizophrenia Than Non-Candidate Genes”, Johnson et al 2017
- “Genome-Wide Association Study Reveals First Locus for Anorexia Nervosa and Metabolic Correlations”, Duncan et al 2016
- “Polygenic Transmission Disequilibrium Confirms That Common and Rare Variation Act Additively to Create Risk for Autism Spectrum Disorders”, Weiner et al 2016
- “Partitioning Heritability Analysis Reveals a Shared Genetic Basis of Brain Anatomy and Schizophrenia”, Lee et al 2016
- “Genetic Overlap between Schizophrenia and Developmental Psychopathology: a Longitudinal Approach Applied to Common Childhood Disorders between Age 7 and 15 Years”, Nivard et al 2016
- “Genetic Evidence for Natural Selection in Humans in the Contemporary United States”, Beauchamp 2016
- “Genome-Wide Analyses of Empathy and Systemizing: Heritability and Correlates With Sex, Education, and Psychiatric Risk”, Warrier et al 2016
- “Molecular Genetic Contributions to Self-Rated Health”, Harris et al 2016
- “Genetic Contributions to Self-Reported Tiredness”, Deary et al 2016
- “Genome-Wide Association Study of Cognitive Functions and Educational Attainment in UK Biobank (n = 112 151)”, Davies et al 2016
- “Individuals With Pronounced Schizotypal Traits Are Particularly Successful in Tickling Themselves”, Lemaitre et al 2016
- “Molecular Genetic Contributions to Social Deprivation and Household Income in UK Biobank (n = 112,151)”, Hill et al 2016
- “Shared Genetic Aetiology between Cognitive Functions and Physical and Mental Health in UK Biobank (n = 112,151) and 24 GWAS Consortia”, Hagenaars et al 2016
- “Schizophrenia Risk Alleles and Neurodevelopmental Outcomes in Childhood: a Population-Based Cohort Study”, Riglin et al 2016
- “Genome-Wide Analysis of over 106 000 Individuals Identifies 9 Neuroticism-Associated Loci”, Smith et al 2016
- “Genetic Markers of Human Evolution Are Enriched in Schizophrenia”, Srinivasan et al 2016
- “Haplotypes of Common SNPs Can Explain Missing Heritability of Complex Diseases”, Bhatia et al 2015
- “Polygenic Risk Scores for Schizophrenia and Bipolar Disorder Predict Creativity”, Power et al 2015
- “Contrasting Regional Architectures of Schizophrenia and Other Complex Diseases Using Fast Variance Components Analysis”, Loh et al 2015
- “An Atlas of Genetic Correlations across Human Diseases and Traits”, Bulik-Sullivan et al 2015
- “Autistic Spectrum Disorder, Attention Deficit Hyperactivity Disorder, and Psychiatric Comorbidities: A Nationwide Study”, Chen et al 2015
- “IQ and Schizophrenia in a Swedish National Sample: Their Causal Relationship and the Interaction of IQ With Genetic Risk”, Kendler et al 2015
- “Bipolar Disorder and Its Relation to Major Psychiatric Disorders: a Family-Based Study in the Swedish Population”, Song et al 2014
- “Does Population Density and Neighborhood Deprivation Predict Schizophrenia? A Nationwide Swedish Family-Based Study of 2.4 Million Individuals”, Sariaslan et al 2014b
- “Uncovering the Hidden Risk Architecture of the Schizophrenias: Confirmation in Three Independent Genome-Wide Association Studies”, Arnedo 2014
- “Molecular Genetic Evidence for Overlap between General Cognitive Ability and Risk for Schizophrenia: a Report from the Cognitive Genomics ConsorTium (COGENT)”, Lencz et al 2014
- “Large-Scale Genomics Unveils the Genetic Architecture of Psychiatric Disorders”, Gratten et al 2014
- “Biological Insights from 108 Schizophrenia-Associated Genetic Loci”
- “Schizophrenia and Cortical Blindness: Protective Effects and Implications for Language”, Leivada & Boeckx 2014
- “The Contribution of de Novo Coding Mutations to Autism Spectrum Disorder”, Iossifov et al 2014
- “Genetic Predisposition to Schizophrenia Associated With Increased Use of Cannabis”, Power et al 2014
- “Polygenic Risk for Schizophrenia Is Associated With Cognitive Change Between Childhood and Old Age”, McIntosh et al 2013
- “Fecundity of Patients With Schizophrenia, Autism, Bipolar Disorder, Depression, Anorexia Nervosa, or Substance Abuse vs Their Unaffected Siblings”, Power et al 2013
- “Identification of Risk Loci With Shared Effects on Five Major Psychiatric Disorders: a Genome-Wide Analysis”
- “Shared Polygenic Contribution between Childhood Attention-Deficit Hyperactivity Disorder and Adult Schizophrenia”, Hamshere et al 2013
- “Genetic Relationship between Five Psychiatric Disorders Estimated from Genome-Wide SNPs”, Lee et al 2013
- “Family History of Schizophrenia and Bipolar Disorder As Risk Factors for Autism”, Sullivan et al 2012
- “CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics”, Malhotra & Sebat 2012
- “Genome-Wide Association Study of Clinical Dimensions of Schizophrenia: Polygenic Effect on Disorganized Symptoms”, Fanous et al 2012
- “Research in China on the Molecular Genetics of Schizophrenia”, Cui & Jiang 2012
- “Large-Scale Genome-Wide Association Analysis of Bipolar Disorder Identifies a New Susceptibility Locus near ODZ4”, Sklar et al 2011
- “Genome-Wide Association Study Identifies Five New Schizophrenia Loci”, Consortium 2011
- “Is Arson the Crime Most Strongly Associated With Psychosis?--A National Case-Control Study of Arson Risk in Schizophrenia and Other Psychoses”, Anwar et al 2011
- “The Truth Wears Off: Is There Something Wrong With the Scientific Method?”, Lehrer 2010
- “False Memory in Schizophrenia Patients With and without Delusions”, Bhatt et al 2010
- “The Role of Genetic Variation in the Causation of Mental Illness: an Evolution-Informed Framework”, Uher 2009
- “Common Polygenic Variation Contributes to Risk of Schizophrenia and Bipolar Disorder”, Consortium 2009
- “Premorbid IQ in Schizophrenia: A Meta-Analytic Review”, Woodberry et al 2008
- “Scholastic Achievement at Age 16 and Risk of Schizophrenia and Other Psychoses: a National Cohort Study”, MacCabe et al 2007
- “Resolving the Paradox of Common, Harmful, Heritable Mental Disorders: Which Evolutionary Genetic Models Work Best?”, Keller & Miller 2006
- “Premorbid Intellectual Functioning in Bipolar Disorder and Schizophrenia: Results From a Cohort Study of Male Conscripts”, Tiihonen et al 2005
- “A Family History Study of Asperger Syndrome”, Ghaziuddin 2005
- “Schizophrenia and Urbanicity: A Major Environmental Influence—Conditional on Genetic Risk”, Krabbendam & Os 2005
- “Nicotine As Therapy”, Powledge 2004
- “Bipolar Disorder, Schizophrenia, and Other Psychotic Disorders in Adults With Childhood Onset AD/HD And/or Autism Spectrum Disorders”, Stahlberg et al 2004
- “A Longitudinal Study of Premorbid IQ Score and Risk of Developing Schizophrenia, Bipolar Disorder, Severe Depression, and Other Non-Affective Psychoses”, Zammit et al 2004
- “The Global Costs of Schizophrenia”, Knapp 2004
- “Theory of Mind and the Role of IQ in Chronic Disorganized Schizophrenia”, Bruene 2003
- “A Population-Based Cohort Study of Premorbid Intellectual, Language, and Behavioral Functioning in Patients With Schizophrenia, Schizoaffective Disorder, and Non-Psychotic Bipolar Disorder”, Reichenberg et al 2002
- “Evidence for Early-Childhood, Pan-Developmental Impairment Specific to Schizophreniform Disorder: Results From a Longitudinal Birth Cohort”, Cannon et al 2002
- “Effects of Family History and Place and Season of Birth on the Risk of Schizophrenia”, Mortensen 1999
- “At Issue: Is Household Crowding a Risk Factor for Schizophrenia and Bipolar Disorder?”, Torrey 1998
- “Educational Level and Hospital Use in Mental Disorders: A Population-Based Study”, Aro et al 1995
- “Viruses, Schizophrenia, and Bipolar Disorder”, Yolken & Torrey 1995
- “An Economic Evaluation of Manic-Depressive Illness—1991”, Wyatt & Henter 1995
- “Confirming Unexpressed Genotypes for Schizophrenia: Risks in the Offspring of Fischer’s Danish Identical and Fraternal Discordant Twins”, Gottesman & Bertelsen 1989
- “Creativity in Manic-Depressives, Cyclothymes, Their Normal Relatives, and Control Subjects”, Richards et al 1988
- “Creativity and Mental Illness: Prevalence Rates in Writers and Their First-Degree Relatives”, Andreasen 1987
- “Offspring of Twin Pairs Discordant for Psychiatric Illness”, Bertelsen & Gottesman 1986
- “An Experience in Submarine Psychiatry”, Serxner 1968
- “Ontology Of Psychiatric Conditions: Tradeoffs And Failures: To What Degree Are Psychiatric Conditions More like Diseases (always Bad) vs. Diverse Neurotypes (potentially Good)?”
- “Genome-Wide Association Study Results for Educational Attainment Aid in Identifying Genetic Heterogeneity of Schizophrenia”
- Sort By Magic
- Wikipedia
- Miscellaneous
- Link Bibliography
See Also
Links
“Illusory Generalizability of Clinical Prediction Models”
“Cat Ownership and Schizophrenia-Related Disorders and Psychotic-Like Experiences: A Systematic Review and Meta-Analysis: Supplement”, McGrath et al 2023
“Cat Ownership and Schizophrenia-Related Disorders and Psychotic-Like Experiences: A Systematic Review and Meta-Analysis”, McGrath et al 2023
“Rare Coding Variants in Schizophrenia-Associated Genes Affect Generalised Cognition in the UK Biobank”, Fenner et al 2023
“Antipsychotic Drug Efficacy Correlates With the Modulation of D1 rather than D2 Receptor-Expressing Striatal Projection Neurons”, Yun et al 2023
“Long-Acting Injectable Second-Generation Antipsychotics vs Placebo and Their Oral Formulations in Acute Schizophrenia: A Systematic Review and Meta-Analysis of Randomized-Controlled-Trials”, Wang et al 2023b
“Longitudinal Follow-Up of Subsequent Psychiatric Comorbidities among Children and Adolescents With Autism Spectrum Disorder”, Yeh et al 2023
“Common and Rare Variant Associations With Latent Traits Underlying Depression, Bipolar Disorder, and Schizophrenia”, Dattani et al 2023
“Relationship of Family Genetic Risk Score With Diagnostic Trajectory in a Swedish National Sample of Incident Cases of Major Depression, Bipolar Disorder, Other Non-Affective Psychosis, and Schizophrenia”, Kendler et al 2023
“Schizophrenia-Associated Somatic Copy-Number Variants from 12,834 Cases Reveal Recurrent NRXN1 and ABCB11 Disruptions”, Maury et al 2023
“What Is the Frequency and Nature of Visual Hallucinations in Non-Clinical Participants?”, Aynsworth et al 2022
What is the frequency and nature of visual hallucinations in non-clinical participants?
“Polygenic Influences Associated With Adolescent Cognitive Skills”, Mitchell et al 2022
Polygenic influences associated with adolescent cognitive skills
“Voice Patterns As Markers of Schizophrenia: Building a Cumulative Generalizable Approach via a Cross-Linguistic and Meta-Analysis Based Investigation”, Parola et al 2022
“Polygenic Architecture of Rare Coding Variation across 400,000 Exomes”, Weiner et al 2022
Polygenic architecture of rare coding variation across 400,000 exomes
“Is an Elevated Family-Genetic Risk for Major Psychiatric Disorders Specific to Creative Occupations?”, Kendler et al 2022b
Is an elevated family-genetic risk for major psychiatric disorders specific to creative occupations?
“Schizophrenia Polygenic Risk Score and Long-Term Success in the Labour Market: A Cohort Study”, Viinikainen et al 2022
Schizophrenia polygenic risk score and long-term success in the labour market: A cohort study
“The Course of General Cognitive Ability in Individuals With Psychotic Disorders”, Jonas et al 2022
The Course of General Cognitive Ability in Individuals With Psychotic Disorders
“Magical Thinking in Individuals With High Polygenic Risk for Schizophrenia but No Non-Affective Psychoses—A General Population Study”, Saarinen et al 2022
“Multi-Omics Analyses Cannot Identify True-Positive Novel Associations from Underpowered Genome-Wide Association Studies of Four Brain-Related Traits”, Baranger et al 2022
“The Relationship of Major Diseases With Childlessness: a Sibling Matched Case-Control and Population Register Study in Finland and Sweden”, Liu et al 2022
“Occurrence of Psychosis and Bipolar Disorder in Adults With Autism: A Systematic Review and Meta-Analysis”, Varcin et al 2022
“Electroconvulsive Therapy”, Espinoza & Kellner 2022
“Genome-Wide Analyses of ADHD Identify 27 Risk Loci, Refine the Genetic Architecture and Implicate Several Cognitive Domains”, Demontis et al 2022
“Neurocognitive Profile of Adolescents With Early-Onset Schizophrenia and Their Unaffected Siblings”, Vyas et al 2022
Neurocognitive profile of adolescents with early-onset schizophrenia and their unaffected siblings
“Interaction Testing and Polygenic Risk Scoring to Estimate the Association of Common Genetic Variants With Treatment Resistance in Schizophrenia”, Pardiñas et al 2022
“Rare Schizophrenia Risk Variant Burden Is Conserved in Diverse Human Populations”, Liu et al 2022
Rare schizophrenia risk variant burden is conserved in diverse human populations
“Association of Schizophrenia Spectrum Disorders and Violence Perpetration in Adults and Adolescents from 15 Countries: A Systematic Review and Meta-Analysis”, Whiting et al 2021
“Role of Polygenic Risk Score in the Familial Transmission of Bipolar Disorder in Youth”, Birmaher et al 2021
Role of Polygenic Risk Score in the Familial Transmission of Bipolar Disorder in Youth
“High-Impact Rare Genetic Variants in Severe Schizophrenia”, Zoghbi et al 2021
“Familial Clustering of Psychiatric Disorders and Low IQ”, Weiser et al 2021
“Deletion of Loss-Of-Function-Intolerant Genes and Risk of 5 Psychiatric Disorders”, Wainberg et al 2021
Deletion of Loss-of-Function-Intolerant Genes and Risk of 5 Psychiatric Disorders
“Comparing Copy Number Variations in a Danish Case Cohort of Individuals With Psychiatric Disorders”, Sánchez et al 2021
Comparing Copy Number Variations in a Danish Case Cohort of Individuals With Psychiatric Disorders
“Novel Disease Associations With Schizophrenia Genetic Risk Revealed in ~400,000 UK Biobank Participants”, Zhang et al 2021b
“A Selection Pressure Landscape for 870 Human Polygenic Traits”, Song et al 2021
A selection pressure landscape for 870 human polygenic traits
“A General Framework for Identifying Rare Variant Combinations in Complex Disorders”, Pounraja & Girirajan 2021
A general framework for identifying rare variant combinations in complex disorders
“Investigating Perceived Heritability of Mental Health Disorders and Attitudes toward Genetic Testing in the United States, United Kingdom, and Australia”, Morosoli et al 2021
“Independent Contribution of Polygenic Risk for Schizophrenia and Cannabis Use in Predicting Psychotic-Like Experiences in Young Adulthood: Testing Gene × Environment Moderation and Mediation”, Elkrief et al 2021
“Adolescent Cannabis Use and Adult Psychoticism: A Longitudinal Co-Twin Control Analysis Using Data from Two Cohorts”, Schaefer et al 2021
“The Prevalence of Mental Disorders among Homeless People in High-Income Countries: An Updated Systematic Review and Meta-Regression Analysis”, Gutwinski et al 2021
“Genome-Wide Association Meta-Analysis of Childhood and Adolescent Internalizing Symptoms”, Jami et al 2021
Genome-wide association meta-analysis of childhood and adolescent internalizing symptoms
“Treating Cognitive Impairment in Schizophrenia With GLP-1RAs: an Overview of Their Therapeutic Potential”, Flintoff et al 2021
“Cognitive Behavior Therapy for Depression From an Evolutionary Perspective”, Hollon et al 2021
Cognitive Behavior Therapy for Depression From an Evolutionary Perspective
“Domestic Mass Shooters: The Association With Unmedicated and Untreated Psychiatric Illness”, Glick et al 2021
Domestic Mass Shooters: The Association With Unmedicated and Untreated Psychiatric Illness
“Career Effects of Mental Health”, Biasi et al 2021 (page 2)
“Utility of Polygenic Embryo Screening for Disease Depends on the Selection Strategy”, Lencz et al 2021
Utility of polygenic embryo screening for disease depends on the selection strategy
“Leroy’s Elusive Little People: A Systematic Review on Lilliputian Hallucinations”, Blom 2021
Leroy’s elusive little people: A systematic review on lilliputian hallucinations
“No Causal Associations between Childhood Family Income and Subsequent Psychiatric Disorders, Substance Misuse and Violent Crime Arrests: a Nationwide Finnish Study of >650 000 Individuals and Their Siblings”, Sariaslan et al 2021
“Ultra-Rare, Rare, and Common Genetic Variant Analysis Converge to Implicate Negative Selection and Neuronal Processes in the Aetiology of Schizophrenia”, Akingbuwa et al 2021
“The Relationship between Cannabis and Schizophrenia: a Genetically Informed Perspective”, Johnson et al 2021
The relationship between cannabis and schizophrenia: a genetically informed perspective
“A Comparison of Ten Polygenic Score Methods for Psychiatric Disorders Applied across Multiple Cohorts”, Ni et al 2021
“Polygenic Risk for Depression, Anxiety and Neuroticism Are Associated With the Severity and Rate of Change in Depressive Symptoms across Adolescence”, Kwong et al 2021
“Ditching Candidate Gene Association Studies: Lessons from Psychiatric Genetics”, Duarte et al 2021
Ditching candidate gene association studies: lessons from psychiatric genetics
“Polygenic Burden Has Broader Impact on Health, Cognition, and Socioeconomic Outcomes Than Most Rare and High-Risk Copy Number Variants”, Saarentaus et al 2021
“From Genotype to Phenotype: Polygenic Prediction of Complex Human Traits”, Raben et al 2021
From Genotype to Phenotype: polygenic prediction of complex human traits
“Antidepressant Response in Major Depressive Disorder: A Genome-Wide Association Study”, Pain et al 2020
Antidepressant Response in Major Depressive Disorder: A Genome-wide Association Study
“Ten Years of Enhancing Neuro-Imaging Genetics through Meta-Analysis: An Overview from the ENIGMA Genetics Working Group”, Medland et al 2020
“The Comorbidity of Schizophrenia Spectrum and Mood Disorders in Autism Spectrum Disorder”, Chien et al 2020
The Comorbidity of Schizophrenia Spectrum and Mood Disorders in Autism Spectrum Disorder
“Use of Genetically Informed Methods to Clarify the Nature of the Association Between Cannabis Use and Risk for Schizophrenia”, Gillespie & Kendler 2020
“Violence and Mental Disorders: a Structured Review of Associations by Individual Diagnoses, Risk Factors, and Risk Assessment”, Whiting et al 2020
“Genetic Architecture of 11 Major Psychiatric Disorders at Biobehavioral, Functional Genomic, and Molecular Genetic Levels of Analysis”, Grotzinger et al 2020
“Exome Sequencing Identifies Rare Coding Variants in 10 Genes Which Confer Substantial Risk for Schizophrenia”, Singh et al 2020
“Common Variants Contribute to Intrinsic Human Brain Functional Networks”, Zhao et al 2020
Common variants contribute to intrinsic human brain functional networks
“Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia”, Consortium et al 2020
Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia
“Novel Ultra-Rare Exonic Variants Identified in a Founder Population Implicate Cadherins in Schizophrenia”, Lencz et al 2020
“A Case of Mirror Image Agnosia and Mirrored Self-Misidentification Syndrome in Schizophrenia without Dementia or Structural Abnormalities”, Rong et al 2020
“The Polygenic Architecture of Schizophrenia—Rethinking Pathogenesis and Nosology”, Smeland et al 2020
The polygenic architecture of schizophrenia—rethinking pathogenesis and nosology
“Risk in Relatives, Heritability, SNP-Based Heritability, and Genetic Correlations in Psychiatric Disorders: A Review”, Baselmans et al 2020
“Obscure and Unknown: Deliriants of the Edgewood Arsenal Human Experiments”, space_crustacean 2020
Obscure and Unknown: Deliriants of the Edgewood Arsenal Human Experiments
“Conditional GWAS Analysis to Identify Disorder-Specific SNPs for Psychiatric Disorders”, Byrne et al 2020
Conditional GWAS analysis to identify disorder-specific SNPs for psychiatric disorders
“Disentangling Selection on Genetically Correlated Polygenic Traits Using Whole-Genome Genealogies”, Stern et al 2020
Disentangling selection on genetically correlated polygenic traits using whole-genome genealogies
“Need to Account for Familial Confounding in Systematic Review and Meta-Analysis of Prenatal Tobacco Smoke Exposure and Schizophrenia”, Quinn et al 2020
“Is Early Blindness Protective of Psychosis or Are We Turning a Blind Eye to the Lack of Statistical Power?”, Jefsen et al 2020
“Genome-Wide Association Study of Creativity Reveals Genetic Overlap With Psychiatric Disorders, Risk Tolerance, and Risky Behaviors”, Li et al 2020c
“Genetics of Schizophrenia in the South African Xhosa”, Gulsuner et al 2020
“Extended Data Figure 2: GWAS Progress over Time. The Relationship of GWAS Associations to Sample-Size Is Shown in This Plot With Selected SCZ GWAS Meta-Analyses of the past 11 Years. The X-Axis Shows Number of Cases. The Y-Axis Shows the Number of Independent Loci Discovered With at Least One Genome-Wide Statistically-Significant Index SNP in the Discovery Meta-Analysis (eg. without Replication Data)...The Slope of ~4 Newly Discovered Loci per 1000 Cases 2013–2019 Increased to a Slope of ~6 With the Latest Sample-Size Increase.”
{kind=link}
“Risk of Subjection to Violence and Perpetration of Violence in Persons With Psychiatric Disorders in Sweden”, Sariaslan et al 2020
“A Large-Scale Genome-Wide Association Study Meta-Analysis of Cannabis Use Disorder”, Johnson et al 2020
A large-scale genome-wide association study meta-analysis of cannabis use disorder
“Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders”, Consortium 2019
Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders
“Genome-Wide Association Studies in Schizophrenia: Recent Advances, Challenges and Future Perspective”, Dennison et al 2019
Genome-wide association studies in schizophrenia: Recent advances, challenges and future perspective
“Genome-Wide Association Study Identifies 49 Common Genetic Variants Associated With Handedness”, Partida et al 2019
Genome-wide association study identifies 49 common genetic variants associated with handedness
“Risk of Non-Affective Psychotic Disorder or Bipolar Disorder in Autism Spectrum Disorder: a Longitudinal Register-Based Study in the Netherlands”, Schalbroeck et al 2019
“Childhood Adoption and Mental Health in Adulthood: The Role of Gene-Environment Correlations and Interactions in the UK Biobank”, Lehto et al 2019
“Unravelling the Genetic Basis of Schizophrenia and Bipolar Disorder With GWAS: A Systematic Review”, Prata et al 2019
Unravelling the genetic basis of schizophrenia and bipolar disorder with GWAS: A systematic review
“Penetrance and Pleiotropy of Polygenic Risk Scores for Schizophrenia in 106,160 Patients across Four Healthcare Systems”, Zheutlin et al 2019
“Evidence That the Association of Childhood Trauma With Psychosis and Related Psychopathology Is Not Explained by Gene-Environment Correlation: A Monozygotic Twin Differences Approach”, Lecei et al 2019
“Psychiatric Comorbidity in Persons With High-Functioning Autism Spectrum Disorders: Findings from a Tertiary Care Neuropsychiatric Hospital”, Nahar et al 2019
“Genome Wide Meta-Analysis Identifies Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders”, Consortium et al 2019
“Phenotypic Annotation: Using Polygenic Scores to Translate Discoveries From Genome-Wide Association Studies From the Top Down”, Belsky & Harden 2019
“Genome-Wide Analysis Reveals Extensive Genetic Overlap between Schizophrenia, Bipolar Disorder, and Intelligence”, Smeland et al 2019
“The past and Future of Novel, Non-Dopamine-2 Receptor Therapeutics for Schizophrenia: A Critical and Comprehensive Review”, Girgis et al 2019
“Common Polygenic Variations for Psychiatric Disorders and Cognition in Relation to Brain Morphology in the General Pediatric Population”, Alemany et al 2019
“Schizophrenia and Bipolar Illness in the Relatives of University Scientists: An Epidemiological Report on the Creativity-Psychopathology Relationship”, Parnas et al 2019
“Schizophrenia Risk and Reproductive Success: a Mendelian Randomization Study”, Lawn et al 2019
Schizophrenia risk and reproductive success: a Mendelian Randomization study
“Schizophrenia Risk Conferred by Protein-Coding de Novo Mutations”, Howrigan et al 2018
Schizophrenia risk conferred by protein-coding de novo mutations
“Using Genetics to Examine a General Liability to Childhood Psychopathology”, Riglin et al 2018
Using genetics to examine a general liability to childhood psychopathology
“The Genetic Relationship between Female Reproductive Traits and Six Psychiatric Disorders”, Ni et al 2018
The genetic relationship between female reproductive traits and six psychiatric disorders
“Glucagon-Like Peptide-1 Receptor Agonists for Antipsychotic-Associated Cardio-Metabolic Risk Factors: A Systematic Review and Individual Participant Data Meta-Analysis”, Siskind et al 2018
“Analysis of Polygenic Score Usage and Performance across Diverse Human Populations”, Duncan et al 2018
Analysis of Polygenic Score Usage and Performance across Diverse Human Populations
“Genetics & the Geography of Health, Behavior, and Attainment”, Belsky et al 2018
Genetics & the Geography of Health, Behavior, and Attainment
“An Evolutionary Compass for Elucidating Selection Mechanisms Shaping Complex Traits”, Uricchio et al 2018
An evolutionary compass for elucidating selection mechanisms shaping complex traits
“Common Genetic Variants Contribute to Risk of Rare Severe Neurodevelopmental Disorders”, Niemi et al 2018
Common genetic variants contribute to risk of rare severe neurodevelopmental disorders
“Evolutionary Perspectives on Genetic and Environmental Risk Factors for Psychiatric Disorders”, Keller 2018
Evolutionary Perspectives on Genetic and Environmental Risk Factors for Psychiatric Disorders
“Genetic Variants Associated With Subjective Well-Being, Depressive Symptoms, and Neuroticism Identified through Genome-Wide Analyses”, Okbay et al 2018
“GWAS in 446,118 European Adults Identifies 78 Genetic Loci for Self-Reported Habitual Sleep Duration Supported by Accelerometer-Derived Estimates”, Dashti et al 2018
“Genome-Wide Association Analyses of Chronotype in 697,828 Individuals Provides New Insights into Circadian Rhythms in Humans and Links to Disease”, Jones et al 2018
“The Genetic Architecture of Schizophrenia, Bipolar Disorder, Obsessive-Compulsive Disorder and Autism Spectrum Disorder”, O’’Connell et al 2018
“Improving Genetic Prediction by Leveraging Genetic Correlations among Human Diseases and Traits”, Maier et al 2018
Improving genetic prediction by leveraging genetic correlations among human diseases and traits
“Association Between Population Density and Genetic Risk for Schizophrenia”, Colodro-Conde et al 2018
Association Between Population Density and Genetic Risk for Schizophrenia
“Common Schizophrenia Alleles Are Enriched in Mutation-Intolerant Genes and in Regions under Strong Background Selection”, Pardiñas et al 2018
“Genome-Wide Association Meta-Analysis in 269,867 Individuals Identifies New Genetic and Functional Links to Intelligence”, Savage et al 2018
“Effects of Latent Toxoplasmosis on Olfactory Functions of Men and Women”, Flegr et al 2017
Effects of latent Toxoplasmosis on olfactory functions of men and women
“Polygenic Prediction of the Phenome, across Ancestry, in Emerging Adulthood”, Docherty et al 2017
Polygenic prediction of the phenome, across ancestry, in emerging adulthood
“Common Risk Variants Identified in Autism Spectrum Disorder”, Grove et al 2017
“The Molecular Genetics of Participation in the Avon Longitudinal Study of Parents and Children”, Taylor et al 2017
The molecular genetics of participation in the Avon Longitudinal Study of Parents and Children
“Genome-Wide Association Study of Social Relationship Satisfaction: Loci and Correlations With Psychiatric Conditions”, Warrier et al 2017
“Different Worlds”, Alexander 2017
“Linkage Disequilibrium-Dependent Architecture of Human Complex Traits Shows Action of Negative Selection”, Gazal et al 2017
“GWAS Meta-Analysis of Neuroticism (n = 449,484) Identifies Novel Genetic Loci and Pathways”, Nagel et al 2017
GWAS Meta-Analysis of Neuroticism (n = 449,484) Identifies Novel Genetic Loci and Pathways
“Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register”, Hilker et al 2017
“Genome-Wide Analysis of Risk-Taking Behavior and Cross-Disorder Genetic Correlations in 116,255 Individuals from the UK Biobank Cohort”, Strawbridge et al 2017
“Genome-Wide Association Study Identifies 30 Loci Associated With Bipolar Disorder”, Stahl et al 2017
Genome-wide association study identifies 30 Loci Associated with Bipolar Disorder
“Genomic Dissection of Bipolar Disorder and Schizophrenia including 28 Subphenotypes”, Ruderfer et al 2017
Genomic dissection of bipolar disorder and schizophrenia including 28 subphenotypes
“Genome-Wide Association Analyses Identify 44 Risk Variants and Refine the Genetic Architecture of Major Depressive Disorder”, Wray et al 2017
“Quantifying the Impact of Rare and Ultra-Rare Coding Variation across the Phenotypic Spectrum”, Ganna et al 2017
Quantifying the impact of rare and ultra-rare coding variation across the phenotypic spectrum
“Psychiatric Vulnerability in Adults With Intellectual Disability and Autism: A Literature Review”, Coli et al 2017
Psychiatric vulnerability in adults with intellectual disability and autism: A literature review
“Genome-Wide Analysis of 113,968 Individuals in UK Biobank Identifies 4 Loci Associated With Mood Instability”, Ward et al 2017
“No Cognitive-Enhancing Effect of GLP-1 Receptor Agonism in Antipsychotic-Treated, Obese Patients With Schizophrenia”, Ishøy et al 2017
“Widespread Signatures of Positive Selection in Common Risk Alleles Associated to Autism Spectrum Disorder”, Polimanti & Gelernter 2017
“Genetic and Environmental Contributions to the Association Between Cannabis Use and Psychotic-Like Experiences in Young Adult Twins”, Nesvåg et al 2017
“The Structure and Measurement of Unusual Sensory Experiences in Different Modalities: The Multi-Modality Unusual Sensory Experiences Questionnaire (MUSEQ)”, Mitchell et al 2017
“No Evidence That Schizophrenia Candidate Genes Are More Associated With Schizophrenia Than Non-Candidate Genes”, Johnson et al 2017
“Genome-Wide Association Study Reveals First Locus for Anorexia Nervosa and Metabolic Correlations”, Duncan et al 2016
Genome-Wide Association Study Reveals First Locus for Anorexia Nervosa and Metabolic Correlations
“Polygenic Transmission Disequilibrium Confirms That Common and Rare Variation Act Additively to Create Risk for Autism Spectrum Disorders”, Weiner et al 2016
“Partitioning Heritability Analysis Reveals a Shared Genetic Basis of Brain Anatomy and Schizophrenia”, Lee et al 2016
Partitioning heritability analysis reveals a shared genetic basis of brain anatomy and schizophrenia
“Genetic Overlap between Schizophrenia and Developmental Psychopathology: a Longitudinal Approach Applied to Common Childhood Disorders between Age 7 and 15 Years”, Nivard et al 2016
“Genetic Evidence for Natural Selection in Humans in the Contemporary United States”, Beauchamp 2016
Genetic evidence for natural selection in humans in the contemporary United States
“Genome-Wide Analyses of Empathy and Systemizing: Heritability and Correlates With Sex, Education, and Psychiatric Risk”, Warrier et al 2016
“Molecular Genetic Contributions to Self-Rated Health”, Harris et al 2016
“Genetic Contributions to Self-Reported Tiredness”, Deary et al 2016
“Genome-Wide Association Study of Cognitive Functions and Educational Attainment in UK Biobank (n = 112 151)”, Davies et al 2016
“Individuals With Pronounced Schizotypal Traits Are Particularly Successful in Tickling Themselves”, Lemaitre et al 2016
Individuals with pronounced schizotypal traits are particularly successful in tickling themselves
“Molecular Genetic Contributions to Social Deprivation and Household Income in UK Biobank (n = 112,151)”, Hill et al 2016
“Shared Genetic Aetiology between Cognitive Functions and Physical and Mental Health in UK Biobank (n = 112,151) and 24 GWAS Consortia”, Hagenaars et al 2016
“Schizophrenia Risk Alleles and Neurodevelopmental Outcomes in Childhood: a Population-Based Cohort Study”, Riglin et al 2016
“Genome-Wide Analysis of over 106 000 Individuals Identifies 9 Neuroticism-Associated Loci”, Smith et al 2016
Genome-wide analysis of over 106 000 individuals identifies 9 neuroticism-associated loci
“Genetic Markers of Human Evolution Are Enriched in Schizophrenia”, Srinivasan et al 2016
Genetic Markers of Human Evolution Are Enriched in Schizophrenia
“Haplotypes of Common SNPs Can Explain Missing Heritability of Complex Diseases”, Bhatia et al 2015
Haplotypes of common SNPs can explain missing heritability of complex diseases
“Polygenic Risk Scores for Schizophrenia and Bipolar Disorder Predict Creativity”, Power et al 2015
Polygenic risk scores for schizophrenia and bipolar disorder predict creativity
“Contrasting Regional Architectures of Schizophrenia and Other Complex Diseases Using Fast Variance Components Analysis”, Loh et al 2015
“An Atlas of Genetic Correlations across Human Diseases and Traits”, Bulik-Sullivan et al 2015
An Atlas of Genetic Correlations across Human Diseases and Traits
“Autistic Spectrum Disorder, Attention Deficit Hyperactivity Disorder, and Psychiatric Comorbidities: A Nationwide Study”, Chen et al 2015
“IQ and Schizophrenia in a Swedish National Sample: Their Causal Relationship and the Interaction of IQ With Genetic Risk”, Kendler et al 2015
“Bipolar Disorder and Its Relation to Major Psychiatric Disorders: a Family-Based Study in the Swedish Population”, Song et al 2014
“Does Population Density and Neighborhood Deprivation Predict Schizophrenia? A Nationwide Swedish Family-Based Study of 2.4 Million Individuals”, Sariaslan et al 2014b
“Uncovering the Hidden Risk Architecture of the Schizophrenias: Confirmation in Three Independent Genome-Wide Association Studies”, Arnedo 2014
“Molecular Genetic Evidence for Overlap between General Cognitive Ability and Risk for Schizophrenia: a Report from the Cognitive Genomics ConsorTium (COGENT)”, Lencz et al 2014
“Large-Scale Genomics Unveils the Genetic Architecture of Psychiatric Disorders”, Gratten et al 2014
Large-scale genomics unveils the genetic architecture of psychiatric disorders
“Biological Insights from 108 Schizophrenia-Associated Genetic Loci”
Biological insights from 108 schizophrenia-associated genetic loci
“Schizophrenia and Cortical Blindness: Protective Effects and Implications for Language”, Leivada & Boeckx 2014
Schizophrenia and cortical blindness: protective effects and implications for language
“The Contribution of de Novo Coding Mutations to Autism Spectrum Disorder”, Iossifov et al 2014
The contribution of de novo coding mutations to autism spectrum disorder
“Genetic Predisposition to Schizophrenia Associated With Increased Use of Cannabis”, Power et al 2014
Genetic predisposition to schizophrenia associated with increased use of cannabis
“Polygenic Risk for Schizophrenia Is Associated With Cognitive Change Between Childhood and Old Age”, McIntosh et al 2013
Polygenic Risk for Schizophrenia Is Associated with Cognitive Change Between Childhood and Old Age
“Fecundity of Patients With Schizophrenia, Autism, Bipolar Disorder, Depression, Anorexia Nervosa, or Substance Abuse vs Their Unaffected Siblings”, Power et al 2013
“Identification of Risk Loci With Shared Effects on Five Major Psychiatric Disorders: a Genome-Wide Analysis”
“Shared Polygenic Contribution between Childhood Attention-Deficit Hyperactivity Disorder and Adult Schizophrenia”, Hamshere et al 2013
“Genetic Relationship between Five Psychiatric Disorders Estimated from Genome-Wide SNPs”, Lee et al 2013
Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs
“Family History of Schizophrenia and Bipolar Disorder As Risk Factors for Autism”, Sullivan et al 2012
Family History of Schizophrenia and Bipolar Disorder as Risk Factors for Autism
“CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics”, Malhotra & Sebat 2012
CNVs: harbingers of a rare variant revolution in psychiatric genetics
“Genome-Wide Association Study of Clinical Dimensions of Schizophrenia: Polygenic Effect on Disorganized Symptoms”, Fanous et al 2012
“Research in China on the Molecular Genetics of Schizophrenia”, Cui & Jiang 2012
Research in China on the molecular genetics of schizophrenia
“Large-Scale Genome-Wide Association Analysis of Bipolar Disorder Identifies a New Susceptibility Locus near ODZ4”, Sklar et al 2011
“Genome-Wide Association Study Identifies Five New Schizophrenia Loci”, Consortium 2011
Genome-wide association study identifies five new schizophrenia loci
“Is Arson the Crime Most Strongly Associated With Psychosis?--A National Case-Control Study of Arson Risk in Schizophrenia and Other Psychoses”, Anwar et al 2011
“The Truth Wears Off: Is There Something Wrong With the Scientific Method?”, Lehrer 2010
The Truth Wears Off: Is there something wrong with the scientific method?
“False Memory in Schizophrenia Patients With and without Delusions”, Bhatt et al 2010
False memory in schizophrenia patients with and without delusions
“The Role of Genetic Variation in the Causation of Mental Illness: an Evolution-Informed Framework”, Uher 2009
The role of genetic variation in the causation of mental illness: an evolution-informed framework
“Common Polygenic Variation Contributes to Risk of Schizophrenia and Bipolar Disorder”, Consortium 2009
Common polygenic variation contributes to risk of schizophrenia and bipolar disorder
“Premorbid IQ in Schizophrenia: A Meta-Analytic Review”, Woodberry et al 2008
“Scholastic Achievement at Age 16 and Risk of Schizophrenia and Other Psychoses: a National Cohort Study”, MacCabe et al 2007
“Resolving the Paradox of Common, Harmful, Heritable Mental Disorders: Which Evolutionary Genetic Models Work Best?”, Keller & Miller 2006
“Premorbid Intellectual Functioning in Bipolar Disorder and Schizophrenia: Results From a Cohort Study of Male Conscripts”, Tiihonen et al 2005
“A Family History Study of Asperger Syndrome”, Ghaziuddin 2005
“Schizophrenia and Urbanicity: A Major Environmental Influence—Conditional on Genetic Risk”, Krabbendam & Os 2005
Schizophrenia and Urbanicity: A Major Environmental Influence—Conditional on Genetic Risk
“Nicotine As Therapy”, Powledge 2004
“Bipolar Disorder, Schizophrenia, and Other Psychotic Disorders in Adults With Childhood Onset AD/HD And/or Autism Spectrum Disorders”, Stahlberg et al 2004
“A Longitudinal Study of Premorbid IQ Score and Risk of Developing Schizophrenia, Bipolar Disorder, Severe Depression, and Other Non-Affective Psychoses”, Zammit et al 2004
“The Global Costs of Schizophrenia”, Knapp 2004
“Theory of Mind and the Role of IQ in Chronic Disorganized Schizophrenia”, Bruene 2003
Theory of mind and the role of IQ in chronic disorganized schizophrenia
“A Population-Based Cohort Study of Premorbid Intellectual, Language, and Behavioral Functioning in Patients With Schizophrenia, Schizoaffective Disorder, and Non-Psychotic Bipolar Disorder”, Reichenberg et al 2002
“Evidence for Early-Childhood, Pan-Developmental Impairment Specific to Schizophreniform Disorder: Results From a Longitudinal Birth Cohort”, Cannon et al 2002
“Effects of Family History and Place and Season of Birth on the Risk of Schizophrenia”, Mortensen 1999
Effects of family history and place and season of birth on the risk of schizophrenia
“At Issue: Is Household Crowding a Risk Factor for Schizophrenia and Bipolar Disorder?”, Torrey 1998
At issue: Is household crowding a risk factor for schizophrenia and bipolar disorder?
“Educational Level and Hospital Use in Mental Disorders: A Population-Based Study”, Aro et al 1995
Educational level and hospital use in mental disorders: A population-based study
“Viruses, Schizophrenia, and Bipolar Disorder”, Yolken & Torrey 1995
“An Economic Evaluation of Manic-Depressive Illness—1991”, Wyatt & Henter 1995
“Confirming Unexpressed Genotypes for Schizophrenia: Risks in the Offspring of Fischer’s Danish Identical and Fraternal Discordant Twins”, Gottesman & Bertelsen 1989
“Creativity in Manic-Depressives, Cyclothymes, Their Normal Relatives, and Control Subjects”, Richards et al 1988
Creativity in manic-depressives, cyclothymes, their normal relatives, and control subjects
“Creativity and Mental Illness: Prevalence Rates in Writers and Their First-Degree Relatives”, Andreasen 1987
Creativity and mental illness: prevalence rates in writers and their first-degree relatives
“Offspring of Twin Pairs Discordant for Psychiatric Illness”, Bertelsen & Gottesman 1986
“An Experience in Submarine Psychiatry”, Serxner 1968
“Ontology Of Psychiatric Conditions: Tradeoffs And Failures: To What Degree Are Psychiatric Conditions More like Diseases (always Bad) vs. Diverse Neurotypes (potentially Good)?”
“Genome-Wide Association Study Results for Educational Attainment Aid in Identifying Genetic Heterogeneity of Schizophrenia”
Sort By Magic
Annotations sorted by machine learning into inferred 'tags'. This provides an alternative way to browse: instead of by date order, one can browse in topic order. The 'sorted' list has been automatically clustered into multiple sections & auto-labeled for easier browsing.
Beginning with the newest annotation, it uses the embedding of each annotation to attempt to create a list of nearest-neighbor annotations, creating a progression of topics. For more details, see the link.
genetic-variation
comorbidity
psychiatric-genetics
neurogenetics
Wikipedia
Miscellaneous
-
/doc/psychiatry/schizophrenia/2021-duarte-figure1-comtphewasnullresults.jpg: -
https://academic.oup.com/schizophreniabulletin/advance-article/doi/10.1093/schbul/sbad173/7517011 -
https://acamh.onlinelibrary.wiley.com/doi/10.1111/jcpp.13528 -
https://aeon.co/essays/a-culture-of-hyper-reality-made-paranoid-delusions-true -
https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.993.7725&rep=rep1&type=pdf -
https://en.wikipedia.org/w/index.php?title=LSD_and_schizophrenia&oldid=699494403 -
https://infoproc.blogspot.com/2014/02/hints-of-genomic-dark-matter-rare.html -
https://link.springer.com/article/10.1007/s00406-023-01654-2 -
https://papers.ssrn.com/sol3/papers.cfm?abstract_id=2544251:View External Link:
-
https://philipdick.com/mirror/websites/pkdweb/short_stories/The%20Days%20Of%20Perky%20Pat.htm -
https://slatestarcodex.com/2016/01/11/schizophrenia-no-smoking-gun/ -
https://slatestarcodex.com/2016/09/12/its-bayes-all-the-way-up/:View External Link:
https://slatestarcodex.com/2016/09/12/its-bayes-all-the-way-up/ -
https://slatestarcodex.com/2017/09/05/book-review-surfing-uncertainty/ -
https://slatestarcodex.com/2018/09/20/treat-the-prodrome/:View External Link:
-
https://slatestarcodex.com/2018/12/11/diametrical-model-of-autism-and-schizophrenia/: -
https://www.damninteresting.com/three-thrown-over-the-cuckoos-nest/ -
https://www.lesswrong.com/posts/bNKcKb8LYaQe49kWF/going-crazy-and-getting-better-again -
https://www.newyorker.com/culture/personal-history/my-brother-toms-schizophrenia -
https://www.newyorker.com/magazine/2023/12/04/what-happens-to-a-school-shooters-sister -
https://www.nytimes.com/2020/04/03/books/review/hidden-valley-road-robert-kolker.html -
https://www.nytimes.com/2023/11/03/well/mind/ozempic-weight-loss-antidepressants-antipsychotics.html -
https://www.salon.com/2012/12/02/better_than_bourne_who_really_killed_nick_deak/:
{kind=link}
{kind=link}
{kind=link}
Link Bibliography
-
2023-mcgrath.pdf: “Cat Ownership and Schizophrenia-Related Disorders and Psychotic-Like Experiences: A Systematic Review and Meta-Analysis”, John J. McGrath, Carmen C. W. Lim, Sukanta Saha -
2022-aynsworth.pdf: “What Is the Frequency and Nature of Visual Hallucinations in Non-Clinical Participants?”, Charlotte Aynsworth, Julie Rolinson, Maryam Pervez, Daniel Collerton, Robert Dudley -
2022-mitchell.pdf: “Polygenic Influences Associated With Adolescent Cognitive Skills”, -
2022-kendler-2.pdf: “Is an Elevated Family-Genetic Risk for Major Psychiatric Disorders Specific to Creative Occupations?”, Kenneth S. Kendler, Henrik Ohlsson, Jan Sundquist, Kristina Sundquist -
2022-jonas.pdf: “The Course of General Cognitive Ability in Individuals With Psychotic Disorders”, -
2021-whiting.pdf: “Association of Schizophrenia Spectrum Disorders and Violence Perpetration in Adults and Adolescents from 15 Countries: A Systematic Review and Meta-Analysis”, Daniel Whiting, Gautam Gulati, John R. Geddes, Seena Fazel -
2021-birmaher.pdf: “Role of Polygenic Risk Score in the Familial Transmission of Bipolar Disorder in Youth”, -
2021-song.pdf: “A Selection Pressure Landscape for 870 Human Polygenic Traits”, Weichen Song, Yueqi Shi, Weidi Wang, Weihao Pan, Wei Qian, Shunying Yu, Min Zhao, Guan Ning Lin -
2021-schaefer.pdf: “Adolescent Cannabis Use and Adult Psychoticism: A Longitudinal Co-Twin Control Analysis Using Data from Two Cohorts”, Jonathan D. Schaefer, Seon-Kyeong Jang, Scott Vrieze, William G. Iacono, Matt McGue, Sylia Wilson -
2021-glick.pdf: “Domestic Mass Shooters: The Association With Unmedicated and Untreated Psychiatric Illness”, Ira D. Glick, Nina E. Cerfolio, Danielle Kamis, Michael J. D. Laurence -
2021-johnson.pdf: “The Relationship between Cannabis and Schizophrenia: a Genetically Informed Perspective”, -
https://www.scielo.br/j/rbp/a/fCXVCnz7PGRpbwNgX6DkJwC/?format=pdf: “Ditching Candidate Gene Association Studies: Lessons from Psychiatric Genetics”, Rodrigo R. R. Duarte, Helena Brentani, Timothy R. Powell -
https://onlinelibrary.wiley.com/doi/full/10.1002/hbm.25311: “Ten Years of Enhancing Neuro-Imaging Genetics through Meta-Analysis: An Overview from the ENIGMA Genetics Working Group”, -
https://slatestarcodex.com/2017/10/02/different-worlds/: “Different Worlds”, Scott Alexander -
https://www.newyorker.com/magazine/2010/12/13/the-truth-wears-off: “The Truth Wears Off: Is There Something Wrong With the Scientific Method?”, Jonah Lehrer -
2009-uher.pdf: “The Role of Genetic Variation in the Causation of Mental Illness: an Evolution-Informed Framework”, Rudolf Uher -
1988-richards.pdf: “Creativity in Manic-Depressives, Cyclothymes, Their Normal Relatives, and Control Subjects”, Ruth Richards, Dennis K. Kinney, Inge Lunde, Maria Benet, Ann P. C. Merzel