- See Also
-
Links
- “Estimated Sustainable Cost-Based Prices for Diabetes Medicines”, Barber et al 2024
- “Association of Semaglutide With Risk of Suicidal Ideation in a Real-World Cohort”, Wang et al 2024
- “Randomized Open-Label Trial of Semaglutide and Dapagliflozin in Patients With Type 2 Diabetes of Different Pathophysiology”, Dwibedi et al 2024
- “Central Glucagon-Like Peptide 1 Receptor Activation Inhibits Toll-Like Receptor Agonist-Induced Inflammation”, Wong et al 2024
- “Oprah Winfrey’s Revelation about Using Weight-Loss Drugs Is a Game-Changer: Here’s Why”, Trepany 2023
- “Substantial Decrease in Alcohol Use Disorder Symptoms Secondary to Semaglutide Therapy for Weight Loss: A Case Series”, Richards et al 2023
- “Comparative Effectiveness of Semaglutide and Tirzepatide for Weight Loss in Adults With Overweight and Obesity in the US: A Real-World Evidence Study”, Rodriguez et al 2023
- “Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes”, Lincoff et al 2023
- “Novo Nordisk Will Stop the Once-Weekly Injectable Semaglutide Kidney Outcomes Trial, FLOW, Based on Interim Analysis”, Nordisk 2023
- “Her Insurance Refused to Pay for Wegovy, So She Sued: Many Employers and Government Programs Won’t Cover Costly Obesity Medications. A Lawsuit Is Challenging One Such Policy”, Robbins 2023
- “Ozempic Is Making People Buy Less Food, Walmart Says”, Case & Banjo 2023
- “Sharon Osbourne Quit Ozempic Because She’s ‘Too Skinny’”, Wickman 2023
- “A Revolution in Obesity Treatment”, Lingvay & Agarwal 2023
- “What Oprah Winfrey Said about Drugs Used for Weight Loss like Ozempic, Mounjaro”, Kindelan 2023
- “Semaglutide in Early Type 1 Diabetes”, Dandona et al 2023
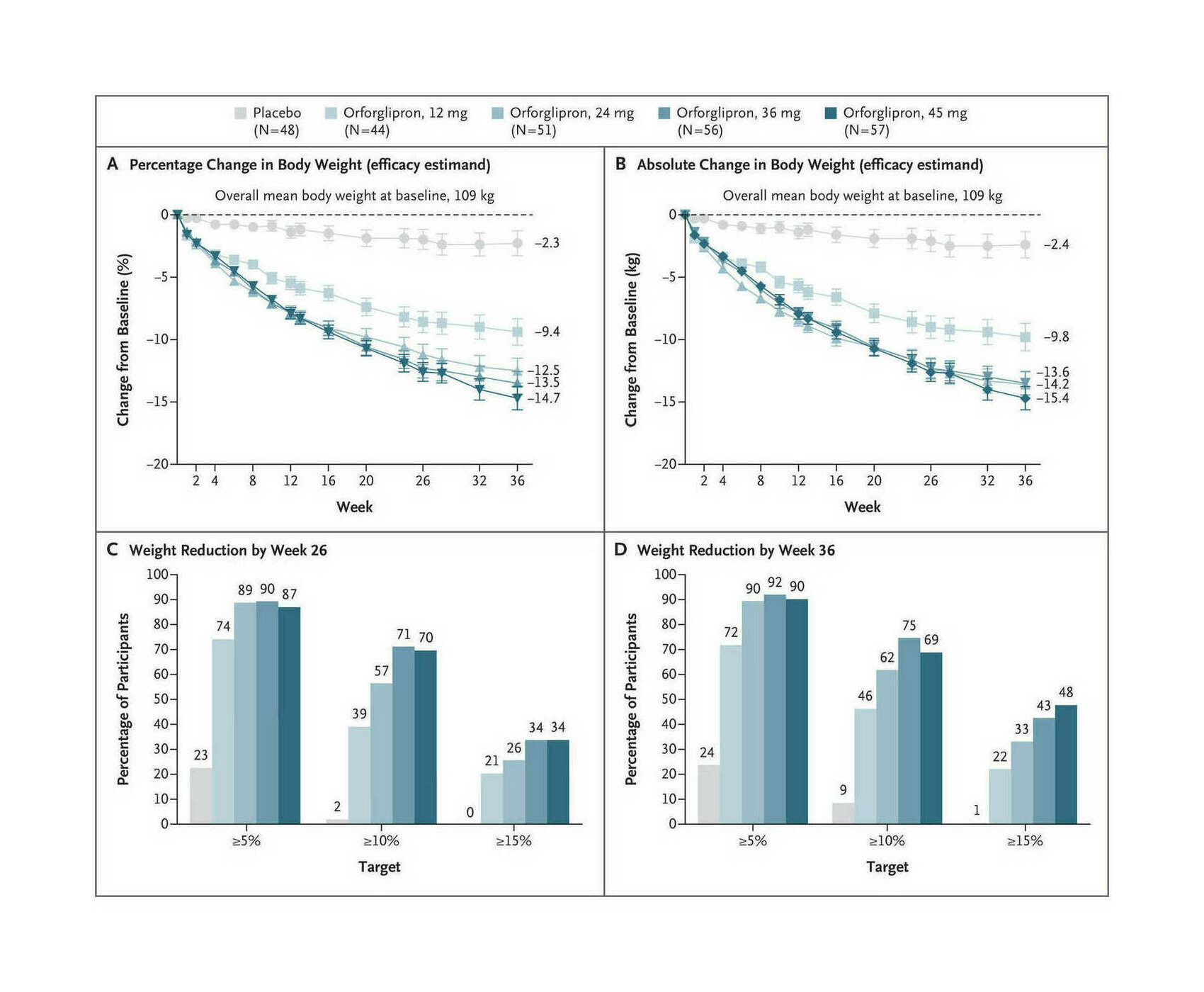
- “Daily Oral GLP-1 Receptor Agonist Orforglipron for Adults With Obesity”, Wharton et al 2023
- “GWAS of Random Glucose in 476,326 Individuals Provide Insights into Diabetes Pathophysiology, Complications and Treatment Stratification”, Lagou et al 2023
- “To Pay for Weight Loss Drugs, Some Take Second Jobs, Ring Up Credit Card Debts: Some People Pay More Than $10,000 a Year Out-Of-Pocket for Ozempic and Mounjaro”, Armour 2023
- “Semaglutide and Cancer: A Systematic Review and Meta-Analysis”, Nagendra et al 2023
- “US Population Eligibility and Estimated Impact of Semaglutide Treatment on Obesity Prevalence and Cardiovascular Disease Events”, Wong et al 2023
- “Novo Boosted As Trial Shows Weight-Loss Wegovy Drug Has [cardiovascular] Medical Benefits”, Fick & Skydsgaard 2023
- “Triple-Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial: Supplement”, Jastreboff 2023
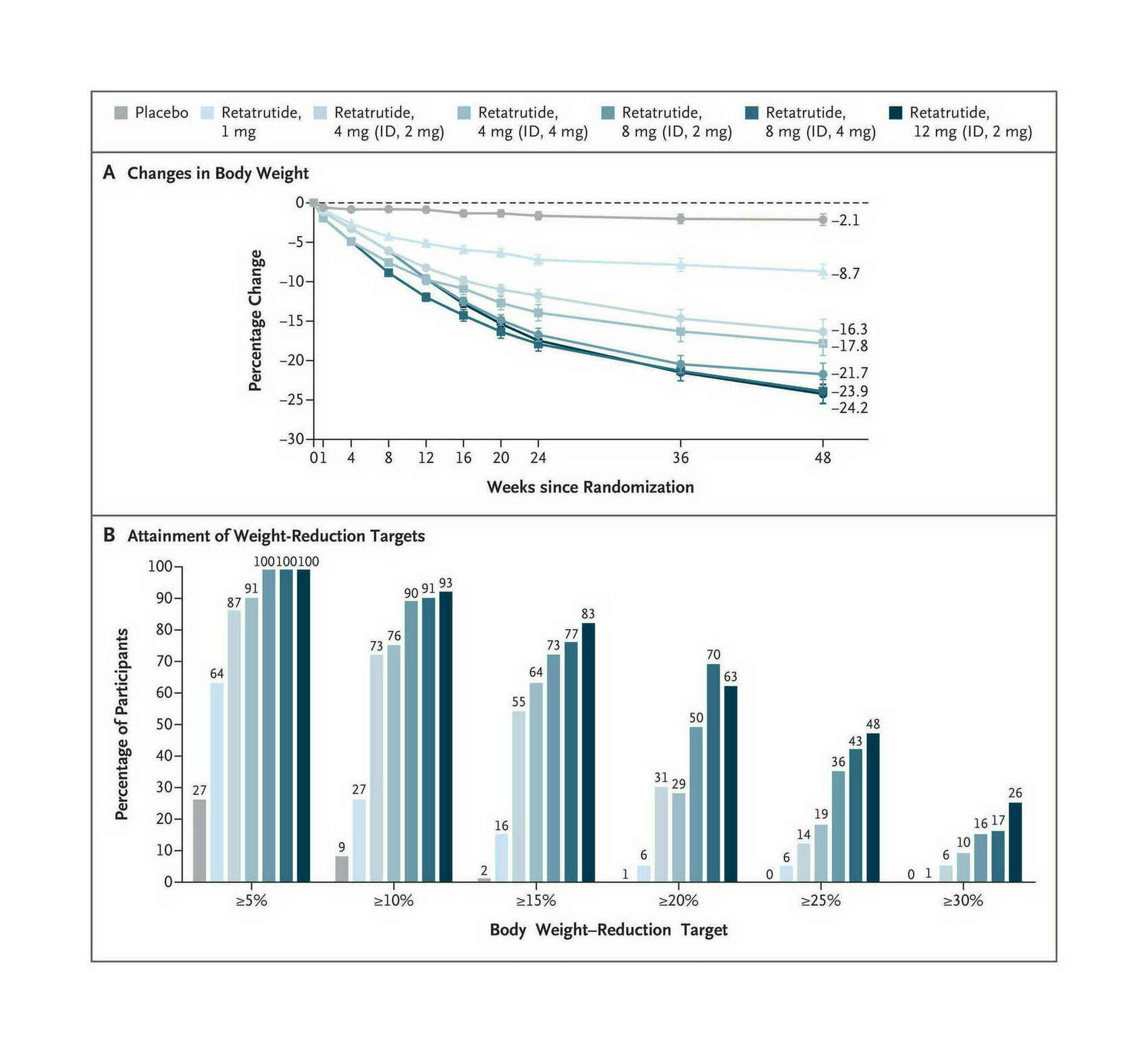
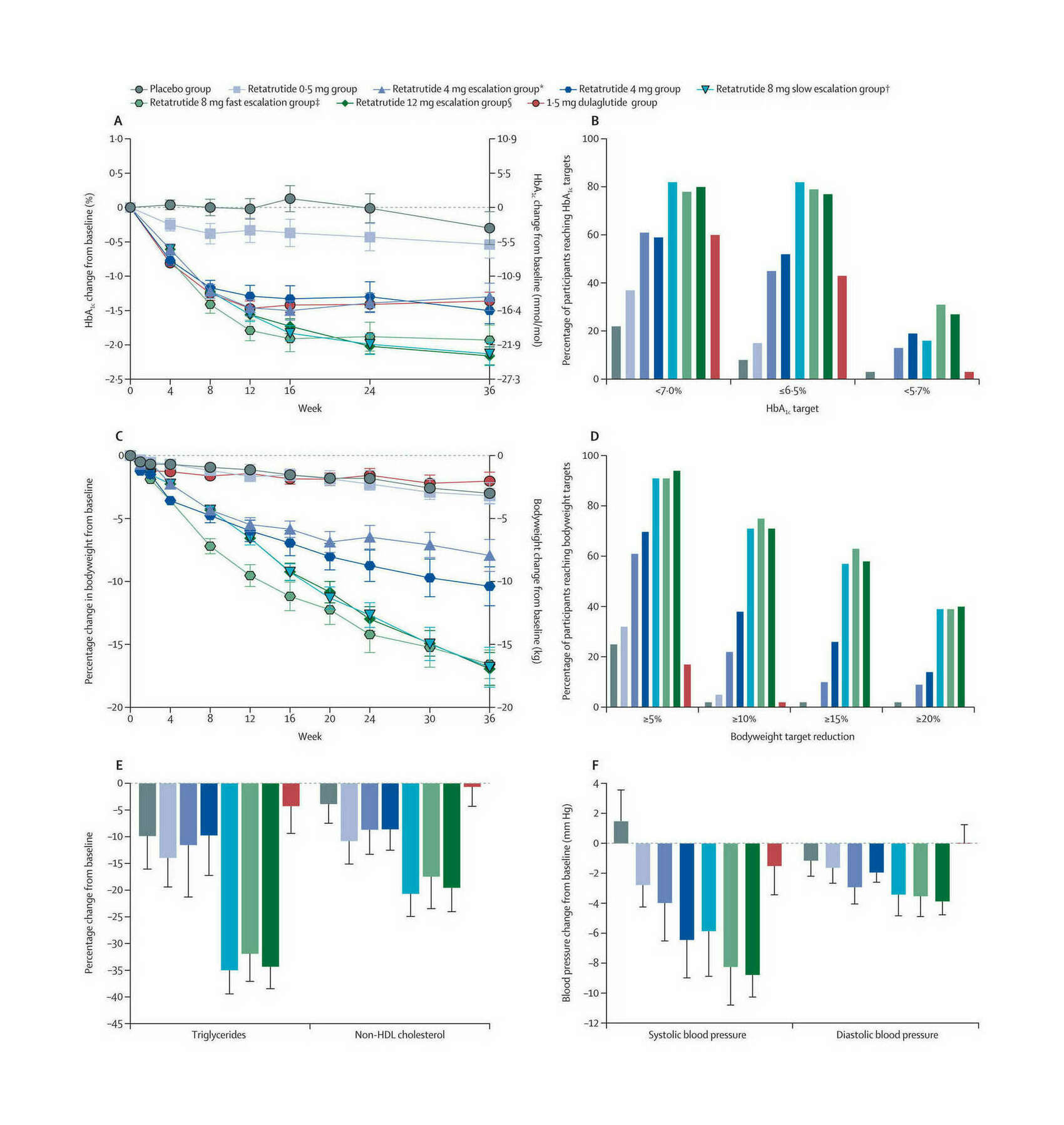
- “Triple-Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial”, Jastreboff et al 2023
- “Retatrutide, a GIP, GLP-1 and Glucagon Receptor Agonist, for People With Type 2 Diabetes: a Randomised, Double-Blind, Placebo and Active-Controlled, Parallel-Group, Phase 2 Trial Conducted in the USA”, Rosenstock et al 2023
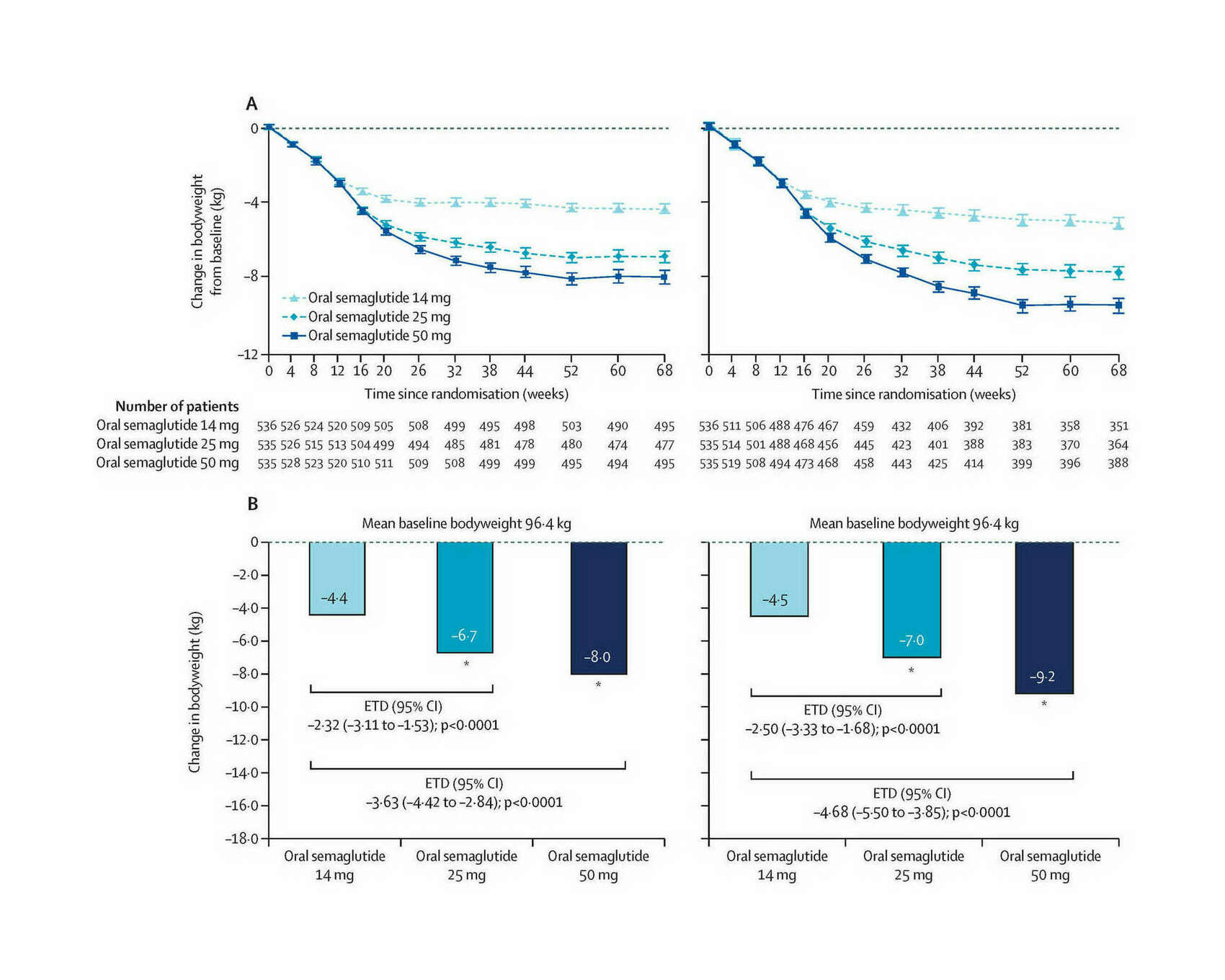
- “Efficacy and Safety of Once-Daily Oral Semaglutide 25 Mg and 50 Mg Compared With 14 Mg in Adults With Type 2 Diabetes (PIONEER PLUS): a Multicentre, Randomised, Phase 3b Trial”, Aroda et al 2023
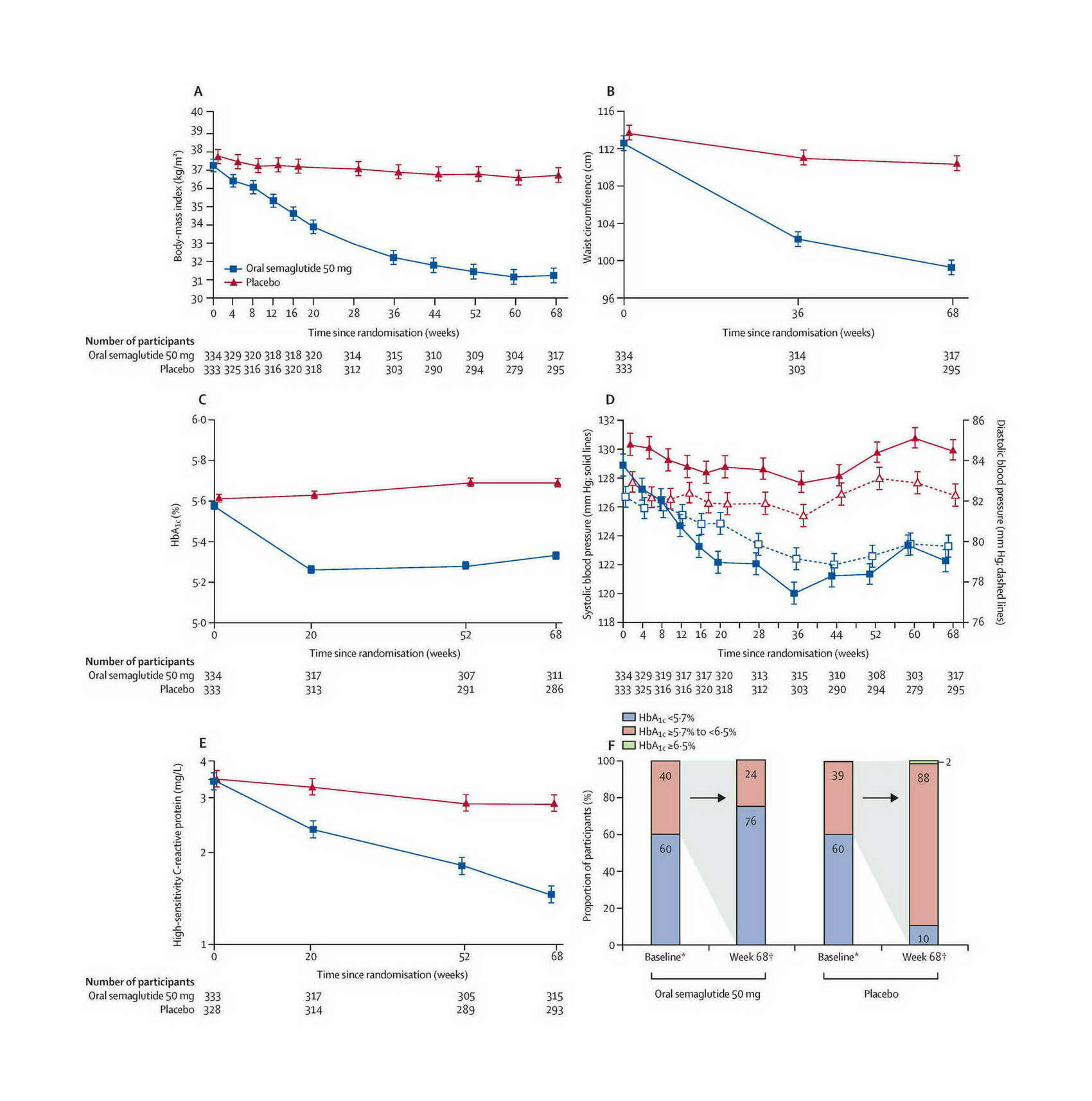
- “Oral Semaglutide 50 Mg Taken Once per Day in Adults With Overweight or Obesity (OASIS 1): a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial”, Knop et al 2023
- “Efficacy and Safety of Co-Administered Once-Weekly Cagrilintide 2.4 Mg With Once-Weekly Semaglutide 2.4 Mg in Type 2 Diabetes: a Multicentre, Randomised, Double-Blind, Active-Controlled, Phase 2 Trial”, Frias et al 2023
- “A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Dose-Finding Study of BI 456906 in People With Overweight/Obesity”, Roux et al 2023
- “Orforglipron (LY3502970), a Novel, Oral Non-Peptide Glucagon-Like Peptide-1 Receptor Agonist: A Phase 1a, Blinded, Placebo-Controlled, Randomized, Single-Dose & Multiple-Ascending-Dose Study in Healthy Participants”, Pratt et al 2023
- “People on Drugs Like Ozempic Say Their ‘Food Noise’ Has Disappeared: For Some, It’s a Startling Side Effect”, Blum 2023
- “What the Scientists Who Pioneered Weight-Loss Drugs Want You to Know”, Reynolds 2023
- “I Lost 40 Pounds on Ozempic. But I’m Left With Even More Questions.”, Marcus 2023
- “Novo Nordisk A/S: Oral Semaglutide 50 Mg Achieved 15.1% Weight Loss in OASIS 1 Trial”, Nordisk 2023
- “Ozempic’s Next Act: People Taking the Drug for Weight Loss Say They Have Also Stopped Drinking, Smoking, Shopping, and Even Nail-Biting”, Zhang 2023
- “Effects of Liraglutide on Depressive Behavior in a Mouse Depression Model and Cognition in the Probe Trial of Morris Water Maze Test”, Seo et al 2023b
- “Feasibility of Exenatide, a GLP-1R Agonist, for Treating Cocaine Use Disorder: A Case Series Study”, Yammine et al 2023
- “A New Drug Switched Off My Appetite. What’s Left? Mounjaro Did What Decades of Struggle With Managing Weight Couldn’t. Welcome to the Post-Hunger Age”, Ford 2023
- “Why Experts Are Urging Swifter Treatment for Children With Obesity: Growing Research Has Shown That Intensive Interventions Are Needed, Scientists Say. Here Is Why Their Advice Is Changing”, Kolata 2023
- “Two-Year Effect of Semaglutide 2.4 Mg on Control of Eating in Adults With Overweight/obesity: STEP 5”, Wharton et al 2023
- “Supplementary Appendix to Aroda Et Al 2032”, Anam 2023
- “Supplement To: Dandona P, Chaudhuri A, Ghanim H. Semaglutide in Early Type 1 Diabetes”, Ghanim 2023
- “Efficacy and Safety of Oral Small Molecule Glucagon-Like Peptide 1 Receptor Agonist Danuglipron for Glycemic Control Among Patients With Type 2 Diabetes: A Randomized Clinical Trial”, Saxena et al 2023
- “Once-Weekly Semaglutide in Adolescents With Obesity”, Weghuber et al 2022
- “Liraglutide Provides Cardioprotection through the Recovery of Mitochondrial Dysfunction and Oxidative Stress in Aging Hearts”, Durak & Turan 2022
- xKloc @ "2022-12-13"
- “Post-Bariatric Patients See More Benefits With Semaglutide vs Liraglutide—Semaglutide Users Also More Likely to Experience Weight Loss, Retrospective Study Suggests”, Dotinga 2022
- “Weight Loss TikTok Trend Triggers Shortage of Diabetic Medication”, Bui 2022
- “Dose Titration With the Glucagon-Like Peptide-1 Agonist, Liraglutide, Reduces Cue- and Drug-Induced Heroin Seeking in High Drug-Taking Rats”, Evans et al 2022
- “Endpoints and Estimands: Understanding Trials of Weight-Loss Drugs”, Adler 2022
- “Two-Year Effects of Semaglutide in Adults With Overweight or Obesity: the STEP 5 Trial”, Garvey et al 2022
- “Hollywood’s Secret New Weight Loss Drug, Revealed: The Hype and Hazards of Ozempic”, Variety 2022
- “Next Generation GLP-1/GIP/glucagon Triple Agonists Normalize Body Weight in Obese Mice”, Knerr et al 2022
- “The Doctor Prescribed an Obesity Drug. Her Insurer Called It ‘Vanity.’ Many Insurance Companies Refuse to Cover New Weight Loss Drugs That Their Doctors Deem Medically Necessary.”, Kolata 2022
- “Weight Regain and Cardiometabolic Effects After Withdrawal of Semaglutide: the STEP 1 Trial Extension”, Wilding et al 2022
- “Treatment With Glucagon-Like Peptide-1 Receptor Agonists and Incidence of Dementia: Data from Pooled Double-Blind Randomized Controlled Trials and Nationwide Disease and Prescription Registers”, Nørgaard et al 2022
- “Effect of Weekly Subcutaneous Semaglutide vs Daily Liraglutide on Body Weight in Adults With Overweight or Obesity Without Diabetes: The STEP 8 Randomized Clinical Trial”, Rubino et al 2022
- “Exenatide Once Weekly for Alcohol Use Disorder Investigated in a Randomized, Placebo-Controlled Clinical Trial”, Klausen et al 2022
- “Once-Weekly Cagrilintide for Weight Management in People With Overweight and Obesity: a Multicentre, Randomized, Double-Blind, Placebo-Controlled and Active-Controlled, Dose-Finding Phase 2 Trial”, Lau et al 2021
- “Anti-Obesity Drug Discovery: Advances and Challenges”, Müller et al 2021
- “Wegovy™ Demonstrated Substantial and Sustained Weight Loss in Two-Year Study in Adults With Obesity”, Nordisk 2021
- “Clinical Pharmacokinetics of Oral Semaglutide: Analyses of Data from Clinical Pharmacology Trials”, Overgaard et al 2021
- “Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Concomitant Administration of Multiple Doses of Cagrilintide With Semaglutide 2·4 Mg for Weight Management: a Randomized, Controlled, Phase 1b Trial”, Enebo et al 2021
- “Healthy Weight Loss Maintenance With Exercise, Liraglutide, or Both Combined”, Lundgren et al 2021
- “The Gut-Brain Axis: Identifying New Therapeutic Approaches for Type 2 Diabetes, Obesity, and Related Disorders”, Richards et al 2021
- “Semaglutide 2.4 Mg Once a Week in Adults With Overweight or Obesity, and Type 2 Diabetes (STEP 2): a Randomised, Double-Blind, Double-Dummy, Placebo-Controlled, Phase 3 Trial”, Davies et al 2021
- “Anti-Interleukin-21 Antibody and Liraglutide for the Preservation of Β-Cell Function in Adults With Recent-Onset Type 1 Diabetes: a Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial”, Herrath et al 2021
- “Effect of Subcutaneous Semaglutide vs Placebo As an Adjunct to Intensive Behavioral Therapy on Body Weight in Adults With Overweight or Obesity: The STEP 3 Randomized Clinical Trial”, Wadden et al 2021
- “Once-Weekly Semaglutide in Adults With Overweight or Obesity”, Wilding et al 2021
- “The Effect of Semaglutide 2.4 Mg Once Weekly on Energy Intake, Appetite, Control of Eating, and Gastric Emptying in Adults With Obesity”, Friedrichsen et al 2021
- “Testing the Effects of the GLP-1 Receptor Agonist Exenatide on Cocaine Self-Administration and Subjective Responses in Humans With Cocaine Use Disorder”, Angarita et al 2021
- “Glucagon-Like Peptide 1 Receptor Agonists and Chronic Lower Respiratory Disease Exacerbations Among Patients With Type 2 Diabetes”, Albogami et al 2021
- “Amylin As a Future Obesity Treatment”, Dehestani et al 2021
- “Efficacy and Safety of AM833 [cagrilintide] for Weight Loss: A Dose-Finding Trial in Adults With Overweight/Obesity [abstract]”, Lau et al 2020
- “GLP-1R Agonists for the Treatment of Obesity: a Patent Review (2015–present)”, Liu et al 2020
- “Semaglutide 2.4 Mg for the Treatment of Obesity: Key Elements of the STEP Trials 1 to 5”, Kushner et al 2020
- “Activation of GLP-1 Receptors Attenuates Oxycodone Taking and Seeking without Compromising the Antinociceptive Effects of Oxycodone in Rats”, Zhang et al 2020
- “Real-World Adherence and Discontinuation of Glucagon-Like Peptide-1 Receptor Agonists Therapy in Type 2 Diabetes Mellitus Patients in the United States”, Weiss et al 2020
- “Can GLP-1 Be a Target for Reward System Related Disorders? A Qualitative Synthesis and Systematic Review Analysis of Studies on Palatable Food, Drugs of Abuse, and Alcohol”, Eren-Yazicioglu et al 2020
- “Management Of Endocrine Disease: Are All GLP-1 Agonists Equal in the Treatment of Type 2 Diabetes?”, Nauck & Meier 2019
- “Comparative Efficacy, Safety, and Cardiovascular Outcomes With Once-Weekly Subcutaneous Semaglutide in the Treatment of Type 2 Diabetes: Insights from the SUSTAIN 1–7 Trials”, Aroda et al 2019
- “FDA Approves First Oral GLP-1 Treatment for Type 2 Diabetes”, FDA 2019
- “PIONEER 1: Randomized Clinical Trial of the Efficacy and Safety of Oral Semaglutide Monotherapy in Comparison With Placebo in Patients With Type 2 Diabetes”, Aroda et al 2019b
- “Oral Semaglutide versus Subcutaneous Liraglutide and Placebo in Type 2 Diabetes (PIONEER 4): a Randomized, Double-Blind, Phase 3a Trial”, Pratley et al 2019
- “Glucagon-Like Peptide-1 Receptors within the Nucleus of the Solitary Tract Regulate Alcohol-Mediated Behaviors in Rodents”, Vallöf et al 2019
- “The Discovery and Development of Liraglutide and Semaglutide”, Knudsen & Lau 2019
- “Beneficial Effects of GLP-1 Agonist in a Male With Compulsive Food-Related Behavior Associated With Autism”, Järvinen et al 2019
- “Effects of Glucagon-Like Peptide 1 Analogs on Alcohol Intake in Alcohol-Preferring Vervet Monkeys”, Thomsen et al 2019
- “Effect of Additional Oral Semaglutide vs Sitagliptin on Glycated Hemoglobin in Adults With Type 2 Diabetes Uncontrolled With Metformin Alone or With Sulfonylurea: The PIONEER 3 Randomized Clinical Trial”, Rosenstock et al 2019
- “The Effect of Glucagon-Like Peptide-1 (GLP-1) Receptor Agonists on Substance Use Disorder (SUD)-Related Behavioral Effects of Drugs and Alcohol: A Systematic Review”, Brunchmann et al 2019
- “Glucagon, GLP-1 and Thermogenesis”, González-García et al 2019
- “Quantifying the Value of Orally Delivered Biologic Therapies: A Cost-Effectiveness Analysis of Oral Semaglutide”, Abramson et al 2019
- “Longer-Term Liraglutide Administration at the Highest Dose Approved for Obesity Increases Reward-Related Orbitofrontal Cortex Activation in Response to Food Cues: Implications for Plateauing Weight Loss in Response to Anti-Obesity Therapies”, Farr et al 2019
- “Glucagon Regulation of Energy Expenditure”, Kleinert et al 2019
- “Safety and Pharmacokinetics of Single and Multiple Ascending Doses of the Novel Oral Human GLP-1 Analogue, Oral Semaglutide, in Healthy Subjects and Subjects With Type 2 Diabetes”, Granhall et al 2018
- “Glucagon-Like Peptide-1 Receptor Agonists for Antipsychotic-Associated Cardio-Metabolic Risk Factors: A Systematic Review and Individual Participant Data Meta-Analysis”, Siskind et al 2018
- “Efficacy and Safety of Semaglutide Compared With Liraglutide and Placebo for Weight Loss in Patients With Obesity: a Randomized, Double-Blind, Placebo and Active Controlled, Dose-Ranging, Phase 2 Trial”, O’Neil et al 2018
- “Liraglutide for Psychiatric Disorders: Clinical Evidence and Challenges”, Camkurt et al 2018
- “Semaglutide versus Dulaglutide Once Weekly in Patients With Type 2 Diabetes (SUSTAIN 7): a Randomised, Open-Label, Phase 3b Trial”, Pratley et al 2018
- “Cost of Achieving HbA1c Treatment Targets and Weight Loss Responses With Once-Weekly Semaglutide Versus Dulaglutide in the United States”, Wilkinson et al 2018
- “Glucagon-Like Peptide-1 Receptor Activation in the Ventral Tegmental Area Attenuates Cocaine Seeking in Rats”, Hernandez et al 2018
- “Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial”, Davies et al 2017
- “Effects of Liraglutide on Weight, Satiation, and Gastric Functions in Obesity: a Randomised, Placebo-Controlled Pilot Trial”, Halawi et al 2017
- “Effects of Once-Weekly Semaglutide on Appetite, Energy Intake, Control of Eating, Food Preference and Body Weight in Subjects With Obesity”, Blundell et al 2017
- “Semaglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes”, Marso et al 2016
- “Unimolecular Polypharmacy for Treatment of Diabetes and Obesity”, Tschöp et al 2016
- “Neurobehavioral Effects of Liraglutide and Sitagliptin in Experimental Models”, Kamble et al 2016
- “A Phase 2, Randomized, Dose-Finding Study of the Novel Once-Weekly Human GLP-1 Analog, Semaglutide, Compared With Placebo and Open-Label Liraglutide in Patients With Type 2 Diabetes”, Nauck et al 2016
- “Glucagon Increases Energy Expenditure Independently of Brown Adipose Tissue Activation in Humans”, Salem et al 2016
- “Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide”, Lau et al 2015
- “Glucagon-Like Peptide-1 (GLP-1) Receptor Agonist Prevents Development of Tolerance to Anti-Anxiety Effect of Ethanol and Withdrawal-Induced Anxiety in Rats”, Sharma et al 2014
- “An Overview of Once-Weekly Glucagon-Like Peptide-1 Receptor Agonists—Available Efficacy and Safety Data and Perspectives for the Future”, Madsbad et al 2011
- “Peripheral and Central GLP-1 Receptor Populations Mediate the Anorectic Effects of Peripherally Administered GLP-1 Receptor Agonists, Liraglutide and Exendin-4”, Kanoski et al 2011
- “Society Is Fixed, Biology Is Mutable”
- Sort By Magic
- Wikipedia
- Miscellaneous
- Link Bibliography
See Also
Links
“Estimated Sustainable Cost-Based Prices for Diabetes Medicines”, Barber et al 2024
Estimated Sustainable Cost-Based Prices for Diabetes Medicines
“Association of Semaglutide With Risk of Suicidal Ideation in a Real-World Cohort”, Wang et al 2024
Association of semaglutide with risk of suicidal ideation in a real-world cohort
“Randomized Open-Label Trial of Semaglutide and Dapagliflozin in Patients With Type 2 Diabetes of Different Pathophysiology”, Dwibedi et al 2024
“Central Glucagon-Like Peptide 1 Receptor Activation Inhibits Toll-Like Receptor Agonist-Induced Inflammation”, Wong et al 2024
“Oprah Winfrey’s Revelation about Using Weight-Loss Drugs Is a Game-Changer: Here’s Why”, Trepany 2023
Oprah Winfrey’s revelation about using weight-loss drugs is a game-changer: Here’s why
“Substantial Decrease in Alcohol Use Disorder Symptoms Secondary to Semaglutide Therapy for Weight Loss: A Case Series”, Richards et al 2023
“Comparative Effectiveness of Semaglutide and Tirzepatide for Weight Loss in Adults With Overweight and Obesity in the US: A Real-World Evidence Study”, Rodriguez et al 2023
“Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes”, Lincoff et al 2023
Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes
“Novo Nordisk Will Stop the Once-Weekly Injectable Semaglutide Kidney Outcomes Trial, FLOW, Based on Interim Analysis”, Nordisk 2023
“Her Insurance Refused to Pay for Wegovy, So She Sued: Many Employers and Government Programs Won’t Cover Costly Obesity Medications. A Lawsuit Is Challenging One Such Policy”, Robbins 2023
“Ozempic Is Making People Buy Less Food, Walmart Says”, Case & Banjo 2023
“Sharon Osbourne Quit Ozempic Because She’s ‘Too Skinny’”, Wickman 2023
“A Revolution in Obesity Treatment”, Lingvay & Agarwal 2023
“What Oprah Winfrey Said about Drugs Used for Weight Loss like Ozempic, Mounjaro”, Kindelan 2023
What Oprah Winfrey said about drugs used for weight loss like Ozempic, Mounjaro
“Semaglutide in Early Type 1 Diabetes”, Dandona et al 2023
“Daily Oral GLP-1 Receptor Agonist Orforglipron for Adults With Obesity”, Wharton et al 2023
Daily Oral GLP-1 Receptor Agonist Orforglipron for Adults with Obesity
“GWAS of Random Glucose in 476,326 Individuals Provide Insights into Diabetes Pathophysiology, Complications and Treatment Stratification”, Lagou et al 2023
“To Pay for Weight Loss Drugs, Some Take Second Jobs, Ring Up Credit Card Debts: Some People Pay More Than $10,000 a Year Out-Of-Pocket for Ozempic and Mounjaro”, Armour 2023
“Semaglutide and Cancer: A Systematic Review and Meta-Analysis”, Nagendra et al 2023
Semaglutide and cancer: A systematic review and meta-analysis
“US Population Eligibility and Estimated Impact of Semaglutide Treatment on Obesity Prevalence and Cardiovascular Disease Events”, Wong et al 2023
“Novo Boosted As Trial Shows Weight-Loss Wegovy Drug Has [cardiovascular] Medical Benefits”, Fick & Skydsgaard 2023
Novo boosted as trial shows weight-loss Wegovy drug has [cardiovascular] medical benefits
“Triple-Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial: Supplement”, Jastreboff 2023
Triple-Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial: Supplement
“Triple-Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial”, Jastreboff et al 2023
Triple-Hormone-Receptor Agonist Retatrutide for Obesity—A Phase 2 Trial
“Retatrutide, a GIP, GLP-1 and Glucagon Receptor Agonist, for People With Type 2 Diabetes: a Randomised, Double-Blind, Placebo and Active-Controlled, Parallel-Group, Phase 2 Trial Conducted in the USA”, Rosenstock et al 2023
“Efficacy and Safety of Once-Daily Oral Semaglutide 25 Mg and 50 Mg Compared With 14 Mg in Adults With Type 2 Diabetes (PIONEER PLUS): a Multicentre, Randomised, Phase 3b Trial”, Aroda et al 2023
“Oral Semaglutide 50 Mg Taken Once per Day in Adults With Overweight or Obesity (OASIS 1): a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial”, Knop et al 2023
“Efficacy and Safety of Co-Administered Once-Weekly Cagrilintide 2.4 Mg With Once-Weekly Semaglutide 2.4 Mg in Type 2 Diabetes: a Multicentre, Randomised, Double-Blind, Active-Controlled, Phase 2 Trial”, Frias et al 2023
“A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Dose-Finding Study of BI 456906 in People With Overweight/Obesity”, Roux et al 2023
“Orforglipron (LY3502970), a Novel, Oral Non-Peptide Glucagon-Like Peptide-1 Receptor Agonist: A Phase 1a, Blinded, Placebo-Controlled, Randomized, Single-Dose & Multiple-Ascending-Dose Study in Healthy Participants”, Pratt et al 2023
“People on Drugs Like Ozempic Say Their ‘Food Noise’ Has Disappeared: For Some, It’s a Startling Side Effect”, Blum 2023
“What the Scientists Who Pioneered Weight-Loss Drugs Want You to Know”, Reynolds 2023
What the Scientists Who Pioneered Weight-Loss Drugs Want You to Know
“I Lost 40 Pounds on Ozempic. But I’m Left With Even More Questions.”, Marcus 2023
I lost 40 pounds on Ozempic. But I’m left with even more questions.
“Novo Nordisk A/S: Oral Semaglutide 50 Mg Achieved 15.1% Weight Loss in OASIS 1 Trial”, Nordisk 2023
Novo Nordisk A/S: Oral semaglutide 50 mg achieved 15.1% weight loss in OASIS 1 trial
“Ozempic’s Next Act: People Taking the Drug for Weight Loss Say They Have Also Stopped Drinking, Smoking, Shopping, and Even Nail-Biting”, Zhang 2023
“Effects of Liraglutide on Depressive Behavior in a Mouse Depression Model and Cognition in the Probe Trial of Morris Water Maze Test”, Seo et al 2023b
“Feasibility of Exenatide, a GLP-1R Agonist, for Treating Cocaine Use Disorder: A Case Series Study”, Yammine et al 2023
Feasibility of Exenatide, a GLP-1R Agonist, for Treating Cocaine Use Disorder: A Case Series Study
“A New Drug Switched Off My Appetite. What’s Left? Mounjaro Did What Decades of Struggle With Managing Weight Couldn’t. Welcome to the Post-Hunger Age”, Ford 2023
“Why Experts Are Urging Swifter Treatment for Children With Obesity: Growing Research Has Shown That Intensive Interventions Are Needed, Scientists Say. Here Is Why Their Advice Is Changing”, Kolata 2023
“Two-Year Effect of Semaglutide 2.4 Mg on Control of Eating in Adults With Overweight/obesity: STEP 5”, Wharton et al 2023
Two-year effect of semaglutide 2.4 mg on control of eating in adults with overweight/obesity: STEP 5
“Supplementary Appendix to Aroda Et Al 2032”, Anam 2023
“Supplement To: Dandona P, Chaudhuri A, Ghanim H. Semaglutide in Early Type 1 Diabetes”, Ghanim 2023
Supplement to: Dandona P, Chaudhuri A, Ghanim H. Semaglutide in early type 1 diabetes
“Efficacy and Safety of Oral Small Molecule Glucagon-Like Peptide 1 Receptor Agonist Danuglipron for Glycemic Control Among Patients With Type 2 Diabetes: A Randomized Clinical Trial”, Saxena et al 2023
“Once-Weekly Semaglutide in Adolescents With Obesity”, Weghuber et al 2022
“Liraglutide Provides Cardioprotection through the Recovery of Mitochondrial Dysfunction and Oxidative Stress in Aging Hearts”, Durak & Turan 2022
xKloc @ "2022-12-13"
“Post-Bariatric Patients See More Benefits With Semaglutide vs Liraglutide—Semaglutide Users Also More Likely to Experience Weight Loss, Retrospective Study Suggests”, Dotinga 2022
“Weight Loss TikTok Trend Triggers Shortage of Diabetic Medication”, Bui 2022
Weight loss TikTok trend triggers shortage of diabetic medication
“Dose Titration With the Glucagon-Like Peptide-1 Agonist, Liraglutide, Reduces Cue- and Drug-Induced Heroin Seeking in High Drug-Taking Rats”, Evans et al 2022
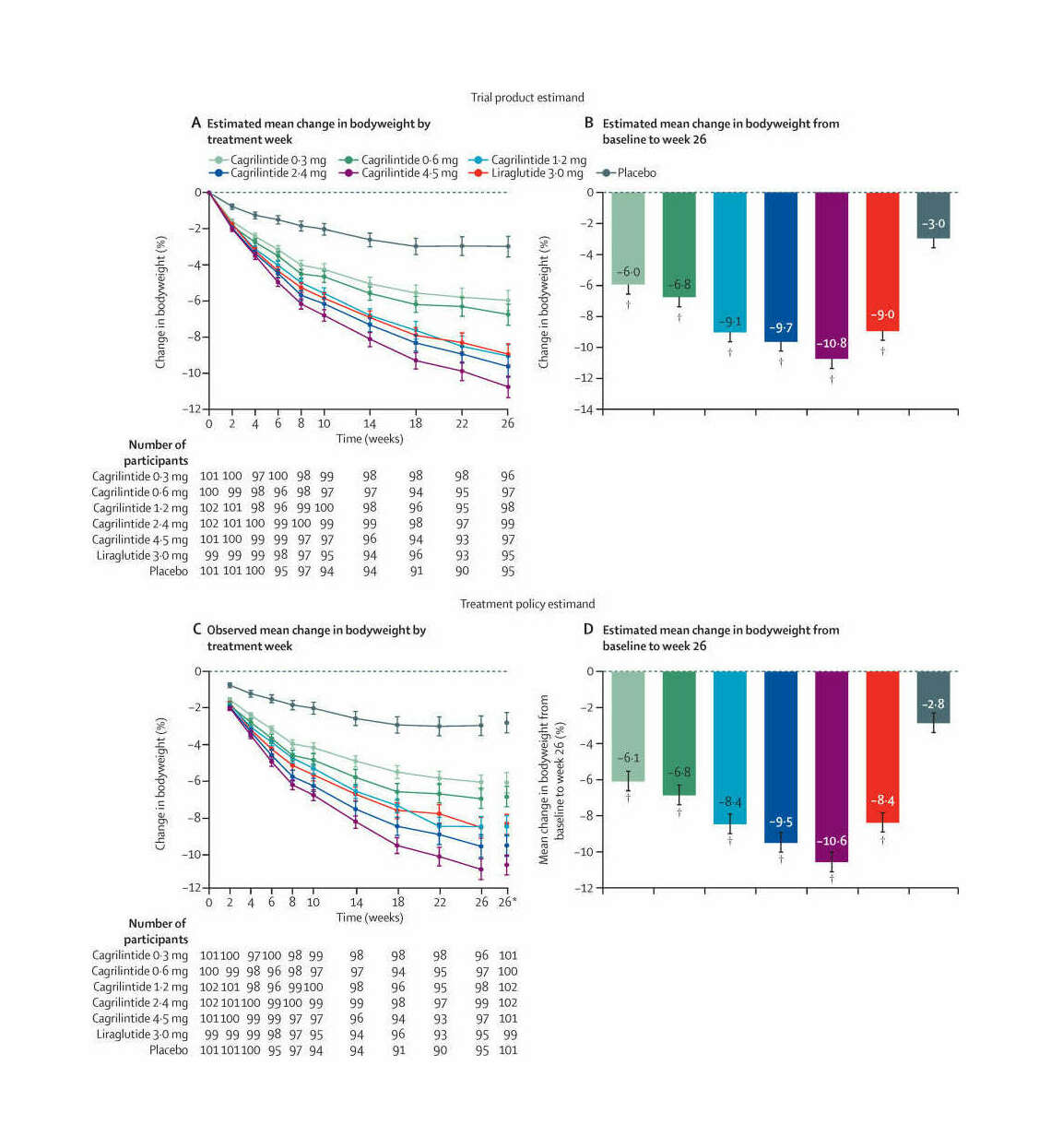
“Endpoints and Estimands: Understanding Trials of Weight-Loss Drugs”, Adler 2022
Endpoints and estimands: understanding trials of weight-loss drugs
“Two-Year Effects of Semaglutide in Adults With Overweight or Obesity: the STEP 5 Trial”, Garvey et al 2022
Two-year effects of semaglutide in adults with overweight or obesity: the STEP 5 trial
“Hollywood’s Secret New Weight Loss Drug, Revealed: The Hype and Hazards of Ozempic”, Variety 2022
Hollywood’s Secret New Weight Loss Drug, Revealed: The Hype and Hazards of Ozempic
“Next Generation GLP-1/GIP/glucagon Triple Agonists Normalize Body Weight in Obese Mice”, Knerr et al 2022
Next generation GLP-1/GIP/glucagon triple agonists normalize body weight in obese mice
“The Doctor Prescribed an Obesity Drug. Her Insurer Called It ‘Vanity.’ Many Insurance Companies Refuse to Cover New Weight Loss Drugs That Their Doctors Deem Medically Necessary.”, Kolata 2022
“Weight Regain and Cardiometabolic Effects After Withdrawal of Semaglutide: the STEP 1 Trial Extension”, Wilding et al 2022
“Treatment With Glucagon-Like Peptide-1 Receptor Agonists and Incidence of Dementia: Data from Pooled Double-Blind Randomized Controlled Trials and Nationwide Disease and Prescription Registers”, Nørgaard et al 2022
“Effect of Weekly Subcutaneous Semaglutide vs Daily Liraglutide on Body Weight in Adults With Overweight or Obesity Without Diabetes: The STEP 8 Randomized Clinical Trial”, Rubino et al 2022
“Exenatide Once Weekly for Alcohol Use Disorder Investigated in a Randomized, Placebo-Controlled Clinical Trial”, Klausen et al 2022
“Once-Weekly Cagrilintide for Weight Management in People With Overweight and Obesity: a Multicentre, Randomized, Double-Blind, Placebo-Controlled and Active-Controlled, Dose-Finding Phase 2 Trial”, Lau et al 2021
“Anti-Obesity Drug Discovery: Advances and Challenges”, Müller et al 2021
“Wegovy™ Demonstrated Substantial and Sustained Weight Loss in Two-Year Study in Adults With Obesity”, Nordisk 2021
Wegovy™ demonstrated substantial and sustained weight loss in two-year study in adults with obesity
“Clinical Pharmacokinetics of Oral Semaglutide: Analyses of Data from Clinical Pharmacology Trials”, Overgaard et al 2021
Clinical Pharmacokinetics of Oral Semaglutide: Analyses of Data from Clinical Pharmacology Trials
“Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Concomitant Administration of Multiple Doses of Cagrilintide With Semaglutide 2·4 Mg for Weight Management: a Randomized, Controlled, Phase 1b Trial”, Enebo et al 2021
“Healthy Weight Loss Maintenance With Exercise, Liraglutide, or Both Combined”, Lundgren et al 2021
Healthy Weight Loss Maintenance with Exercise, Liraglutide, or Both Combined
“The Gut-Brain Axis: Identifying New Therapeutic Approaches for Type 2 Diabetes, Obesity, and Related Disorders”, Richards et al 2021
“Semaglutide 2.4 Mg Once a Week in Adults With Overweight or Obesity, and Type 2 Diabetes (STEP 2): a Randomised, Double-Blind, Double-Dummy, Placebo-Controlled, Phase 3 Trial”, Davies et al 2021
“Anti-Interleukin-21 Antibody and Liraglutide for the Preservation of Β-Cell Function in Adults With Recent-Onset Type 1 Diabetes: a Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial”, Herrath et al 2021
“Effect of Subcutaneous Semaglutide vs Placebo As an Adjunct to Intensive Behavioral Therapy on Body Weight in Adults With Overweight or Obesity: The STEP 3 Randomized Clinical Trial”, Wadden et al 2021
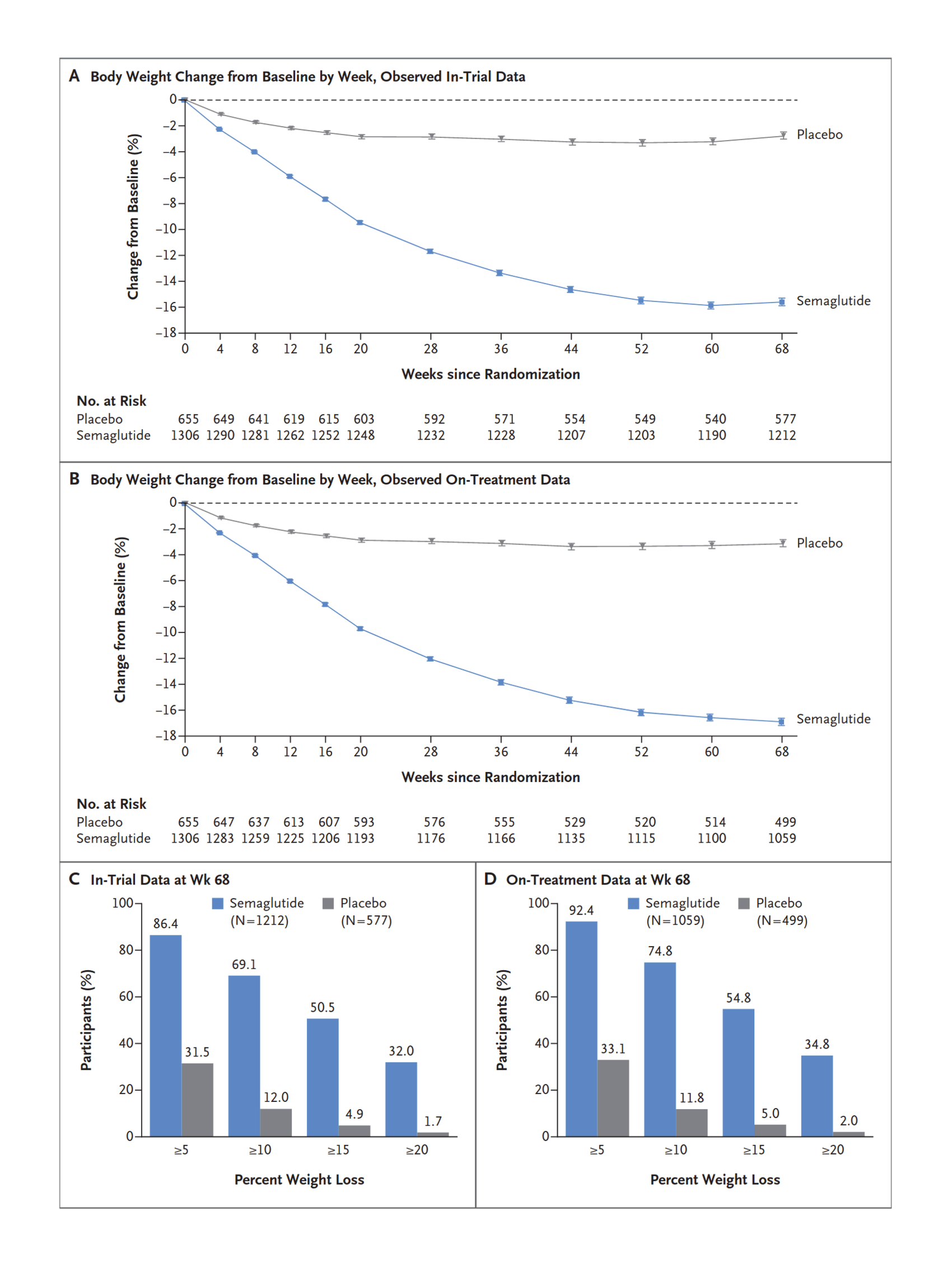
“Once-Weekly Semaglutide in Adults With Overweight or Obesity”, Wilding et al 2021
Once-Weekly Semaglutide in Adults with Overweight or Obesity
“The Effect of Semaglutide 2.4 Mg Once Weekly on Energy Intake, Appetite, Control of Eating, and Gastric Emptying in Adults With Obesity”, Friedrichsen et al 2021
“Testing the Effects of the GLP-1 Receptor Agonist Exenatide on Cocaine Self-Administration and Subjective Responses in Humans With Cocaine Use Disorder”, Angarita et al 2021
“Glucagon-Like Peptide 1 Receptor Agonists and Chronic Lower Respiratory Disease Exacerbations Among Patients With Type 2 Diabetes”, Albogami et al 2021
“Amylin As a Future Obesity Treatment”, Dehestani et al 2021
“Efficacy and Safety of AM833 [cagrilintide] for Weight Loss: A Dose-Finding Trial in Adults With Overweight/Obesity [abstract]”, Lau et al 2020
“GLP-1R Agonists for the Treatment of Obesity: a Patent Review (2015–present)”, Liu et al 2020
GLP-1R agonists for the treatment of obesity: a patent review (2015–present)
“Semaglutide 2.4 Mg for the Treatment of Obesity: Key Elements of the STEP Trials 1 to 5”, Kushner et al 2020
Semaglutide 2.4 mg for the Treatment of Obesity: Key Elements of the STEP Trials 1 to 5
“Activation of GLP-1 Receptors Attenuates Oxycodone Taking and Seeking without Compromising the Antinociceptive Effects of Oxycodone in Rats”, Zhang et al 2020
“Real-World Adherence and Discontinuation of Glucagon-Like Peptide-1 Receptor Agonists Therapy in Type 2 Diabetes Mellitus Patients in the United States”, Weiss et al 2020
“Can GLP-1 Be a Target for Reward System Related Disorders? A Qualitative Synthesis and Systematic Review Analysis of Studies on Palatable Food, Drugs of Abuse, and Alcohol”, Eren-Yazicioglu et al 2020
“Management Of Endocrine Disease: Are All GLP-1 Agonists Equal in the Treatment of Type 2 Diabetes?”, Nauck & Meier 2019
Management Of Endocrine Disease: Are all GLP-1 agonists equal in the treatment of type 2 diabetes?
“Comparative Efficacy, Safety, and Cardiovascular Outcomes With Once-Weekly Subcutaneous Semaglutide in the Treatment of Type 2 Diabetes: Insights from the SUSTAIN 1–7 Trials”, Aroda et al 2019
“FDA Approves First Oral GLP-1 Treatment for Type 2 Diabetes”, FDA 2019
“PIONEER 1: Randomized Clinical Trial of the Efficacy and Safety of Oral Semaglutide Monotherapy in Comparison With Placebo in Patients With Type 2 Diabetes”, Aroda et al 2019b
“Oral Semaglutide versus Subcutaneous Liraglutide and Placebo in Type 2 Diabetes (PIONEER 4): a Randomized, Double-Blind, Phase 3a Trial”, Pratley et al 2019
“Glucagon-Like Peptide-1 Receptors within the Nucleus of the Solitary Tract Regulate Alcohol-Mediated Behaviors in Rodents”, Vallöf et al 2019
“The Discovery and Development of Liraglutide and Semaglutide”, Knudsen & Lau 2019
The Discovery and Development of Liraglutide and Semaglutide
“Beneficial Effects of GLP-1 Agonist in a Male With Compulsive Food-Related Behavior Associated With Autism”, Järvinen et al 2019
“Effects of Glucagon-Like Peptide 1 Analogs on Alcohol Intake in Alcohol-Preferring Vervet Monkeys”, Thomsen et al 2019
Effects of glucagon-like peptide 1 analogs on alcohol intake in alcohol-preferring vervet monkeys
“Effect of Additional Oral Semaglutide vs Sitagliptin on Glycated Hemoglobin in Adults With Type 2 Diabetes Uncontrolled With Metformin Alone or With Sulfonylurea: The PIONEER 3 Randomized Clinical Trial”, Rosenstock et al 2019
“The Effect of Glucagon-Like Peptide-1 (GLP-1) Receptor Agonists on Substance Use Disorder (SUD)-Related Behavioral Effects of Drugs and Alcohol: A Systematic Review”, Brunchmann et al 2019
“Glucagon, GLP-1 and Thermogenesis”, González-García et al 2019
“Quantifying the Value of Orally Delivered Biologic Therapies: A Cost-Effectiveness Analysis of Oral Semaglutide”, Abramson et al 2019
“Longer-Term Liraglutide Administration at the Highest Dose Approved for Obesity Increases Reward-Related Orbitofrontal Cortex Activation in Response to Food Cues: Implications for Plateauing Weight Loss in Response to Anti-Obesity Therapies”, Farr et al 2019
“Glucagon Regulation of Energy Expenditure”, Kleinert et al 2019
“Safety and Pharmacokinetics of Single and Multiple Ascending Doses of the Novel Oral Human GLP-1 Analogue, Oral Semaglutide, in Healthy Subjects and Subjects With Type 2 Diabetes”, Granhall et al 2018
“Glucagon-Like Peptide-1 Receptor Agonists for Antipsychotic-Associated Cardio-Metabolic Risk Factors: A Systematic Review and Individual Participant Data Meta-Analysis”, Siskind et al 2018
“Efficacy and Safety of Semaglutide Compared With Liraglutide and Placebo for Weight Loss in Patients With Obesity: a Randomized, Double-Blind, Placebo and Active Controlled, Dose-Ranging, Phase 2 Trial”, O’Neil et al 2018
“Liraglutide for Psychiatric Disorders: Clinical Evidence and Challenges”, Camkurt et al 2018
Liraglutide for psychiatric disorders: clinical evidence and challenges
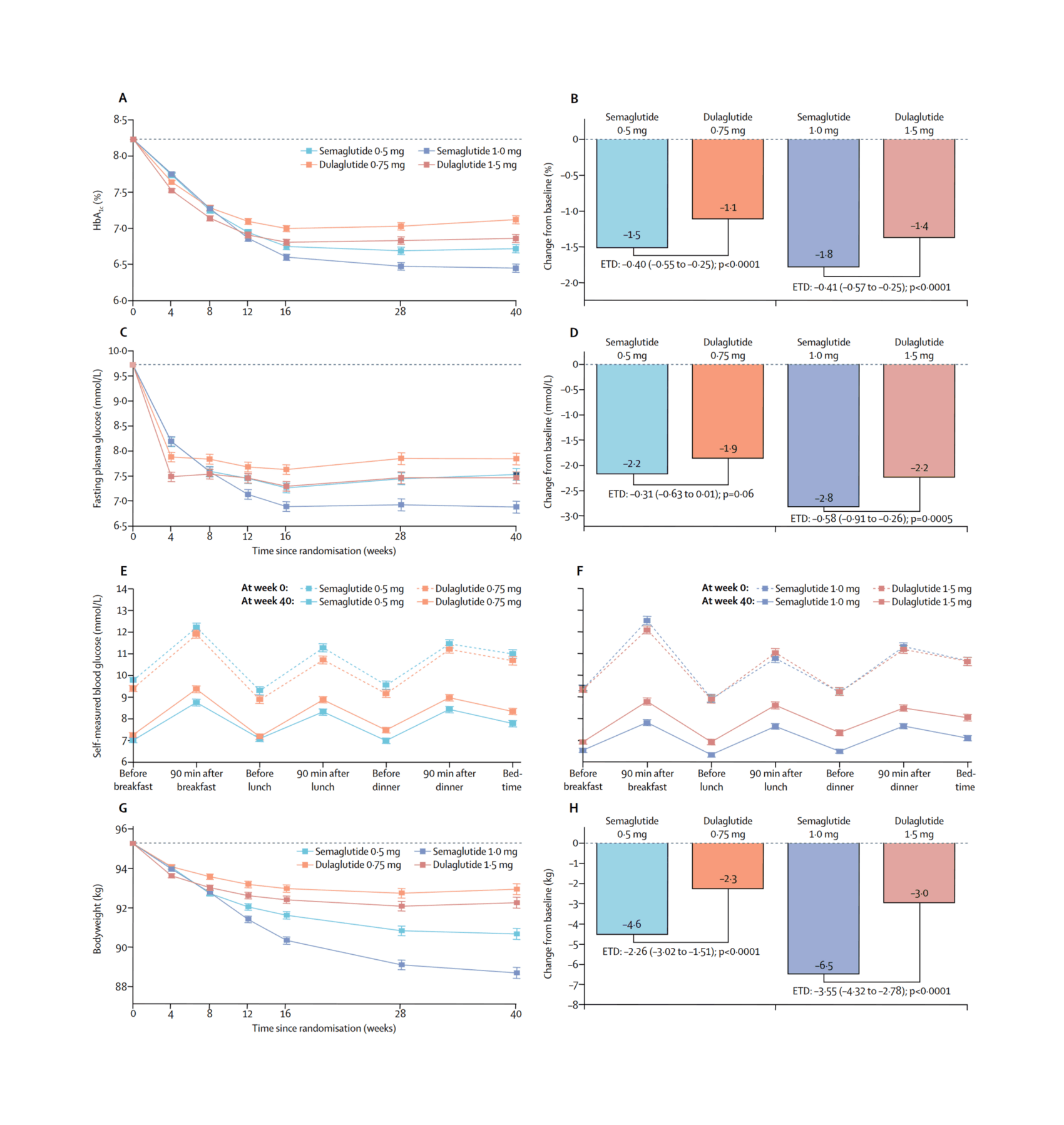
“Semaglutide versus Dulaglutide Once Weekly in Patients With Type 2 Diabetes (SUSTAIN 7): a Randomised, Open-Label, Phase 3b Trial”, Pratley et al 2018
“Cost of Achieving HbA1c Treatment Targets and Weight Loss Responses With Once-Weekly Semaglutide Versus Dulaglutide in the United States”, Wilkinson et al 2018
“Glucagon-Like Peptide-1 Receptor Activation in the Ventral Tegmental Area Attenuates Cocaine Seeking in Rats”, Hernandez et al 2018
“Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial”, Davies et al 2017
“Effects of Liraglutide on Weight, Satiation, and Gastric Functions in Obesity: a Randomised, Placebo-Controlled Pilot Trial”, Halawi et al 2017
“Effects of Once-Weekly Semaglutide on Appetite, Energy Intake, Control of Eating, Food Preference and Body Weight in Subjects With Obesity”, Blundell et al 2017
“Semaglutide and Cardiovascular Outcomes in Patients With Type 2 Diabetes”, Marso et al 2016
Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes
“Unimolecular Polypharmacy for Treatment of Diabetes and Obesity”, Tschöp et al 2016
Unimolecular Polypharmacy for Treatment of Diabetes and Obesity
“Neurobehavioral Effects of Liraglutide and Sitagliptin in Experimental Models”, Kamble et al 2016
Neurobehavioral effects of liraglutide and sitagliptin in experimental models
“A Phase 2, Randomized, Dose-Finding Study of the Novel Once-Weekly Human GLP-1 Analog, Semaglutide, Compared With Placebo and Open-Label Liraglutide in Patients With Type 2 Diabetes”, Nauck et al 2016
“Glucagon Increases Energy Expenditure Independently of Brown Adipose Tissue Activation in Humans”, Salem et al 2016
Glucagon increases energy expenditure independently of brown adipose tissue activation in humans
“Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide”, Lau et al 2015
Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide
“An Overview of Once-Weekly Glucagon-Like Peptide-1 Receptor Agonists—Available Efficacy and Safety Data and Perspectives for the Future”, Madsbad et al 2011
“Peripheral and Central GLP-1 Receptor Populations Mediate the Anorectic Effects of Peripherally Administered GLP-1 Receptor Agonists, Liraglutide and Exendin-4”, Kanoski et al 2011
“Society Is Fixed, Biology Is Mutable”
Sort By Magic
Annotations sorted by machine learning into inferred 'tags'. This provides an alternative way to browse: instead of by date order, one can browse in topic order. The 'sorted' list has been automatically clustered into multiple sections & auto-labeled for easier browsing.
Beginning with the newest annotation, it uses the embedding of each annotation to attempt to create a list of nearest-neighbor annotations, creating a progression of topics. For more details, see the link.
glp-1-therapy
weight-management
semaglutide-effects
anti-obesity
obesity-semaglutide
step-studies
glp-1-agonists
Wikipedia
Miscellaneous
-
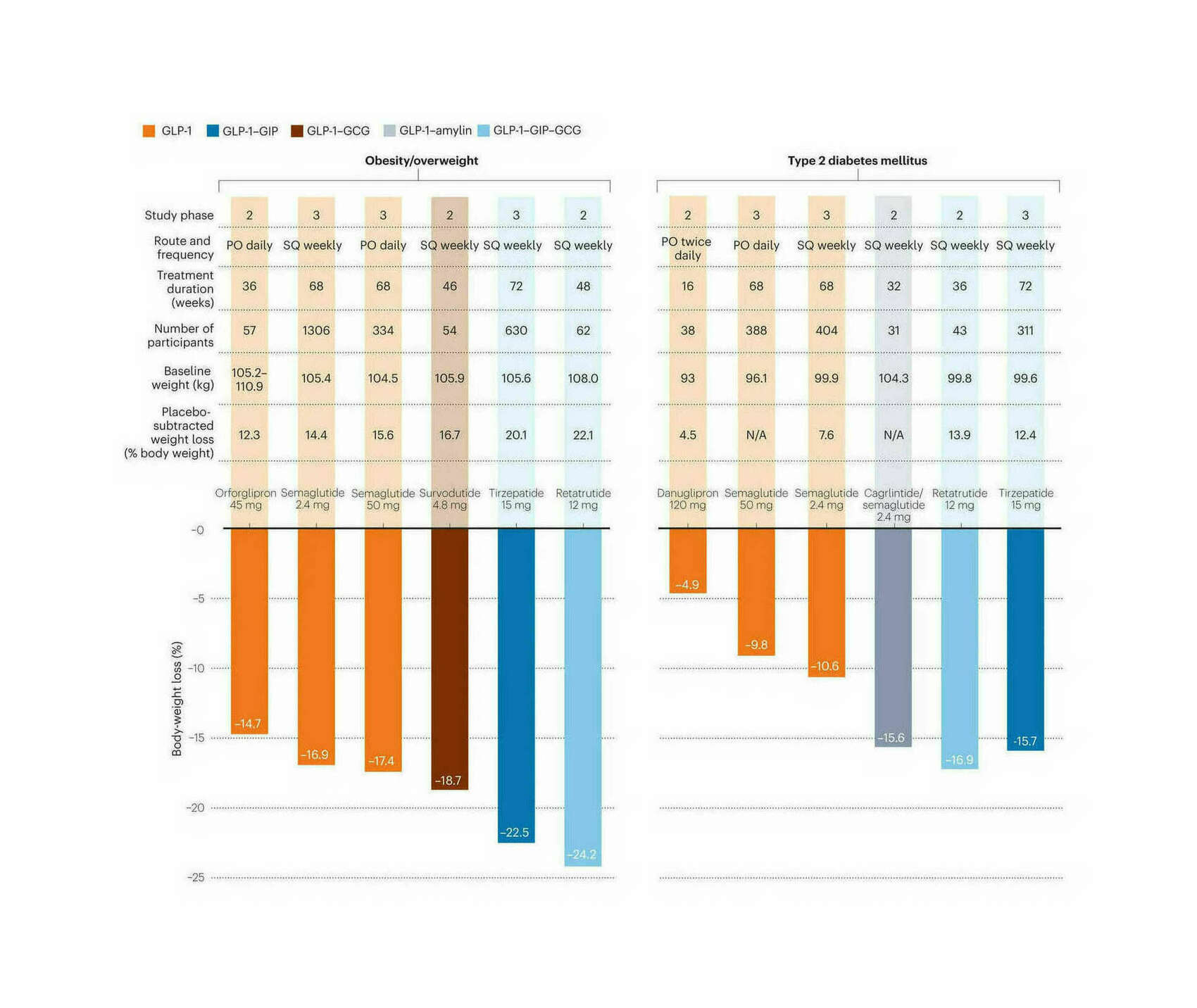
/doc/longevity/glp/semaglutide/2023-lingvay-figure1-comparisonofmajorglp1agonistsforweightloss.jpg: -
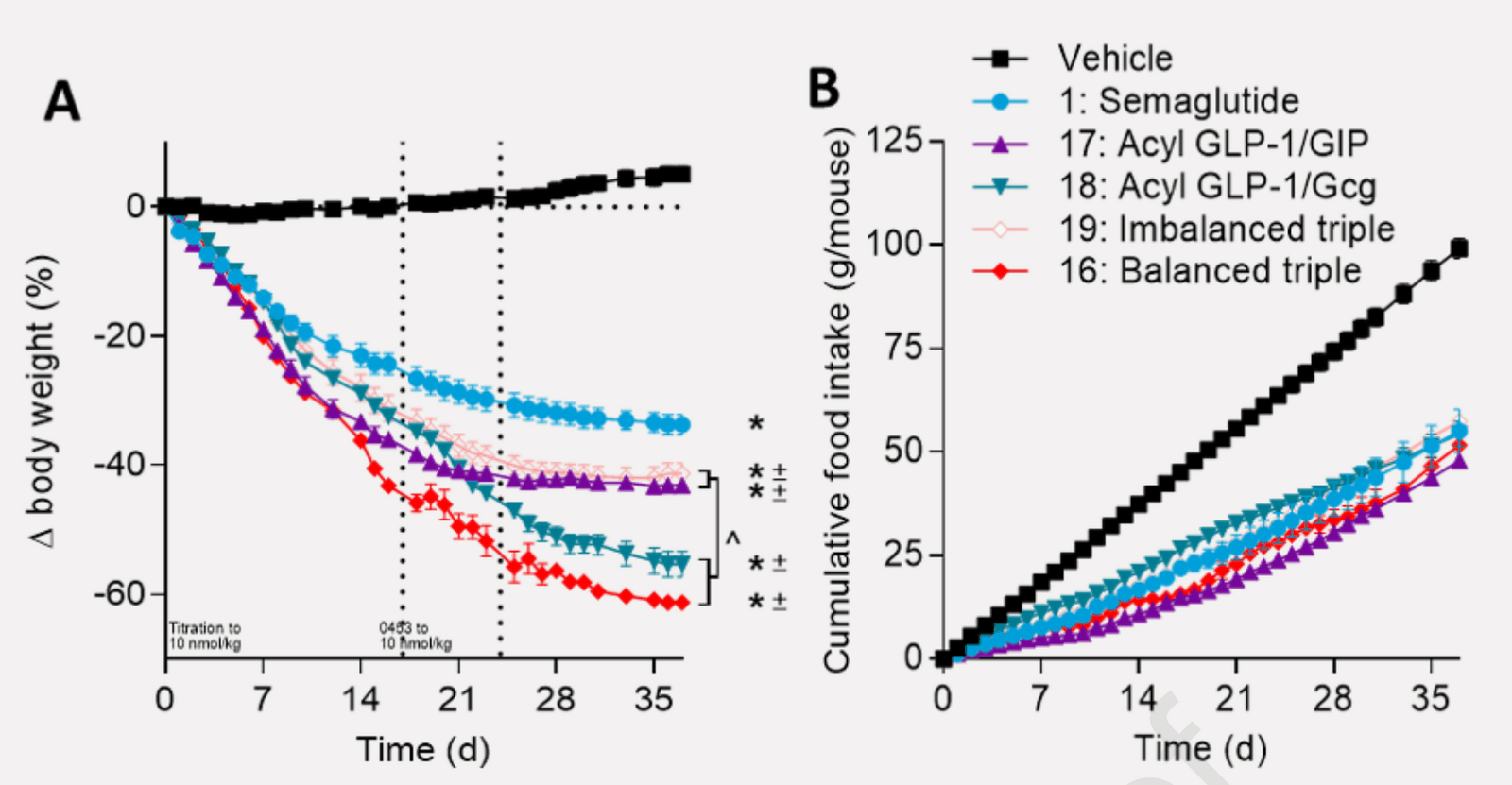
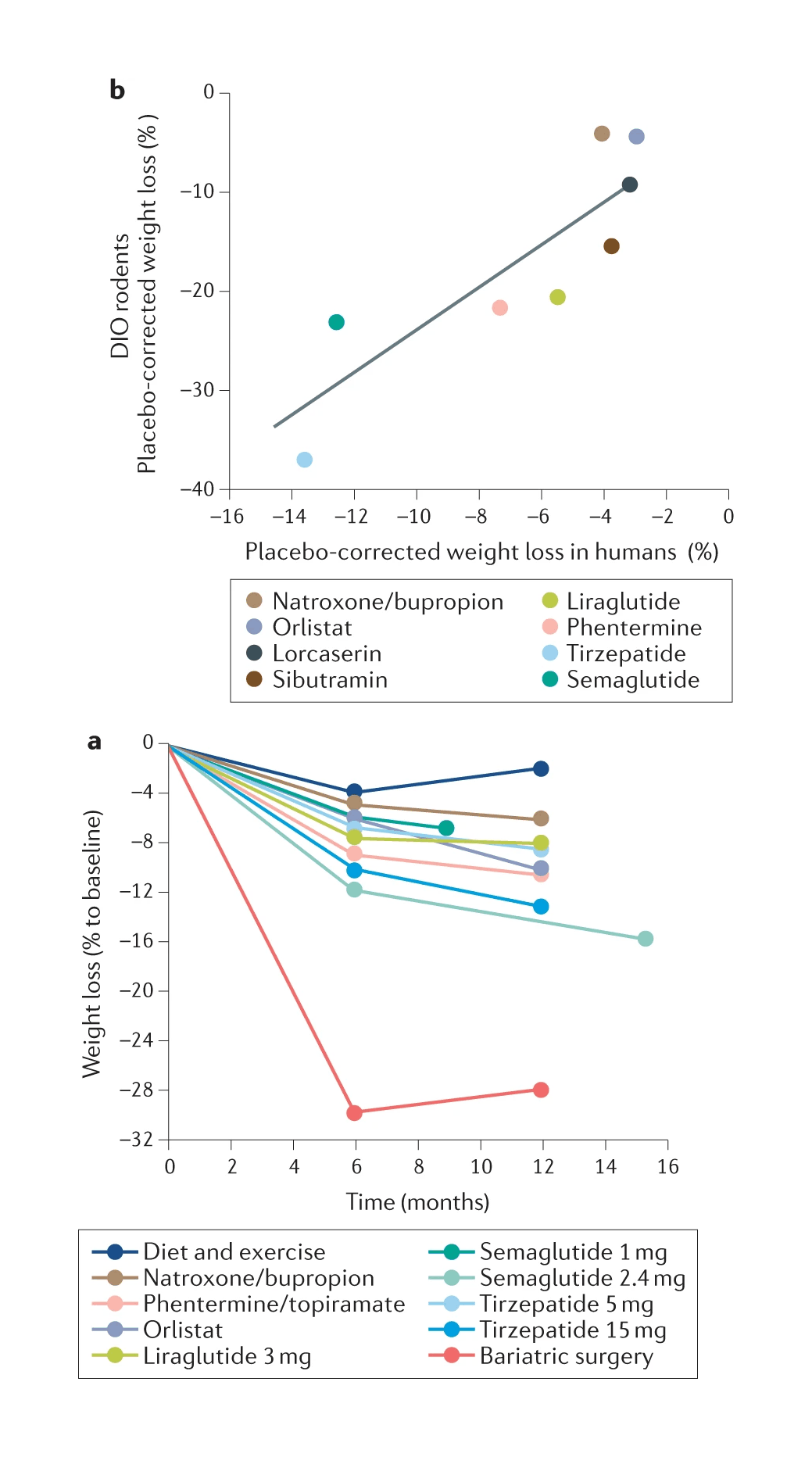
/doc/longevity/glp/semaglutide/2022-knerr-figure2-combinedpeptideweightlosseffectsinmice.png: -
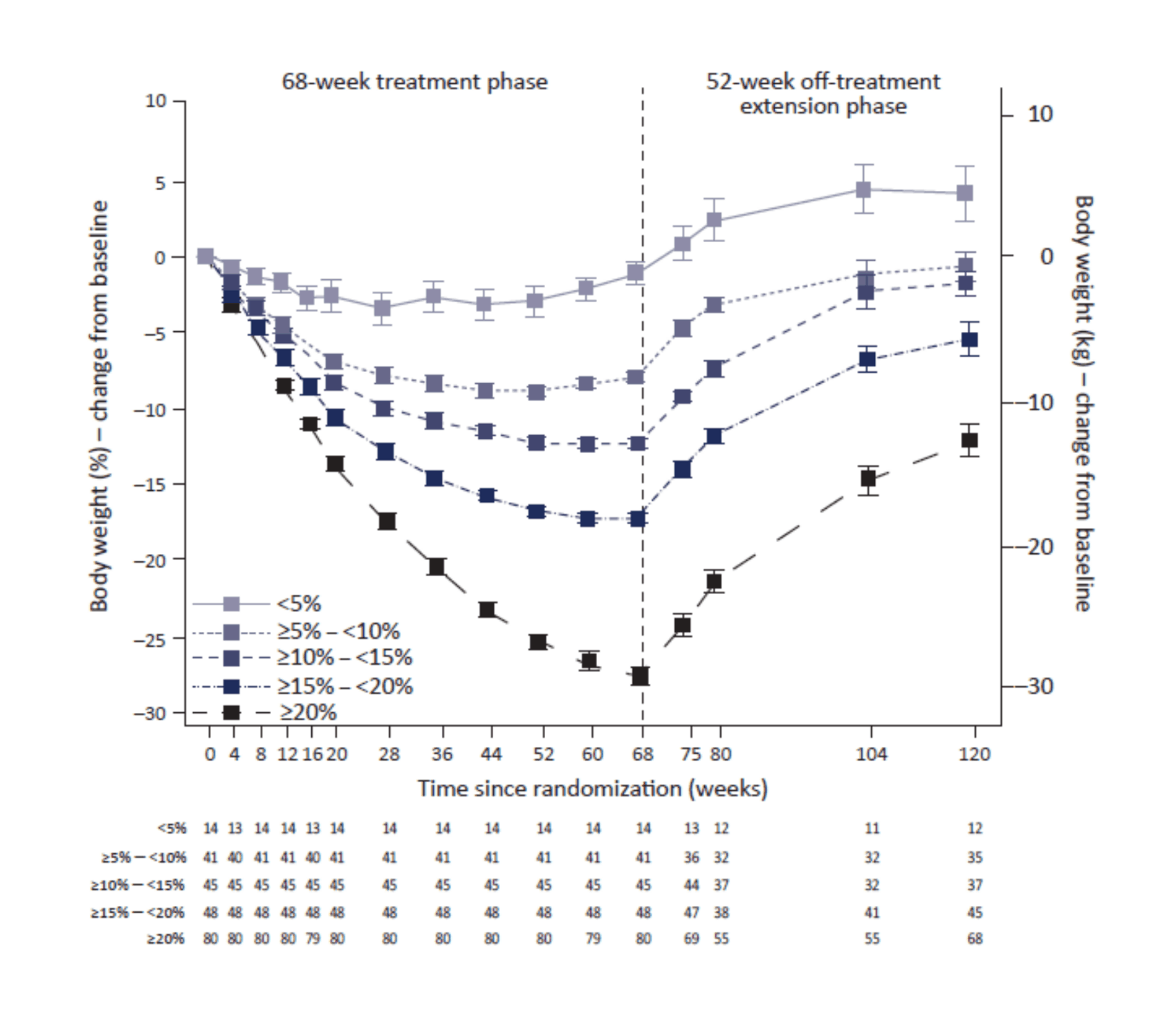
/doc/longevity/glp/semaglutide/2022-wilding-figure1b-regainofbodyweightafterstoppingsemaglutide.png: -
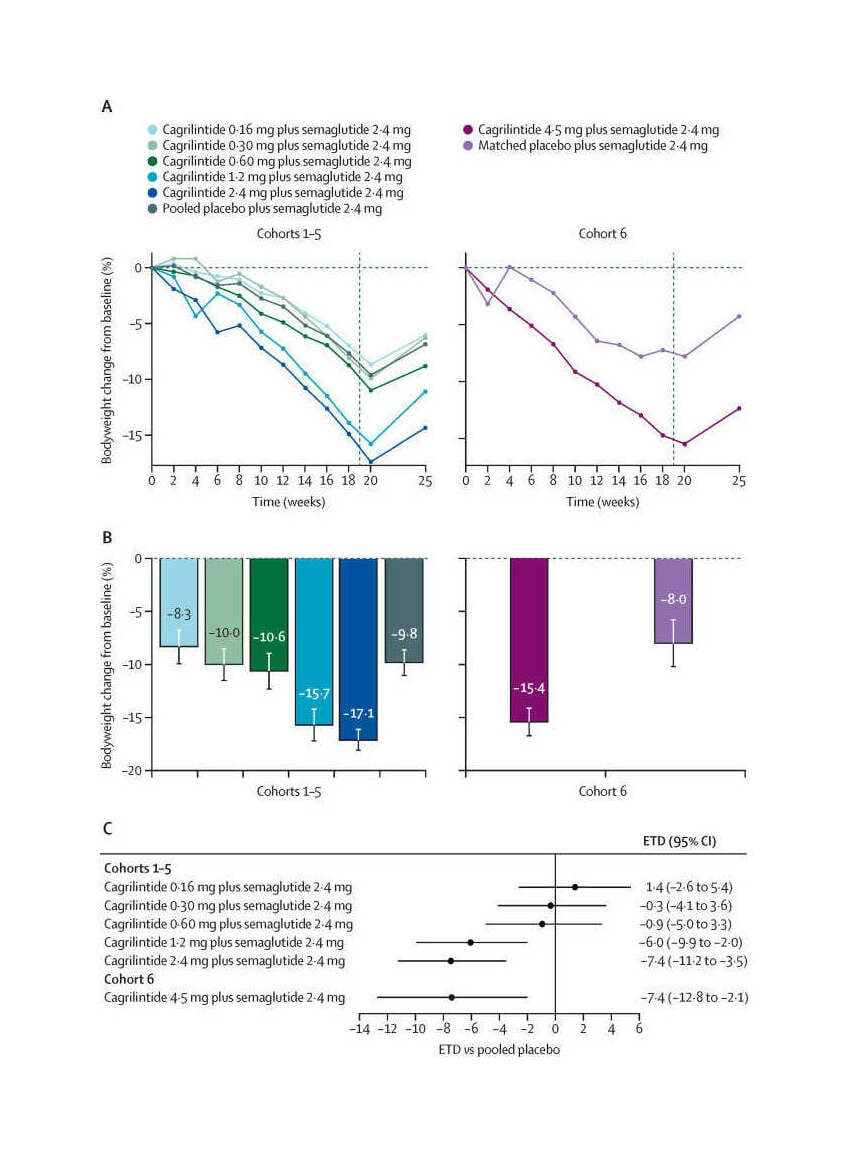
/doc/longevity/glp/semaglutide/2021-lau-figure2-cagrilintidetotalbodyweightlossbydoseovertime.jpg: -
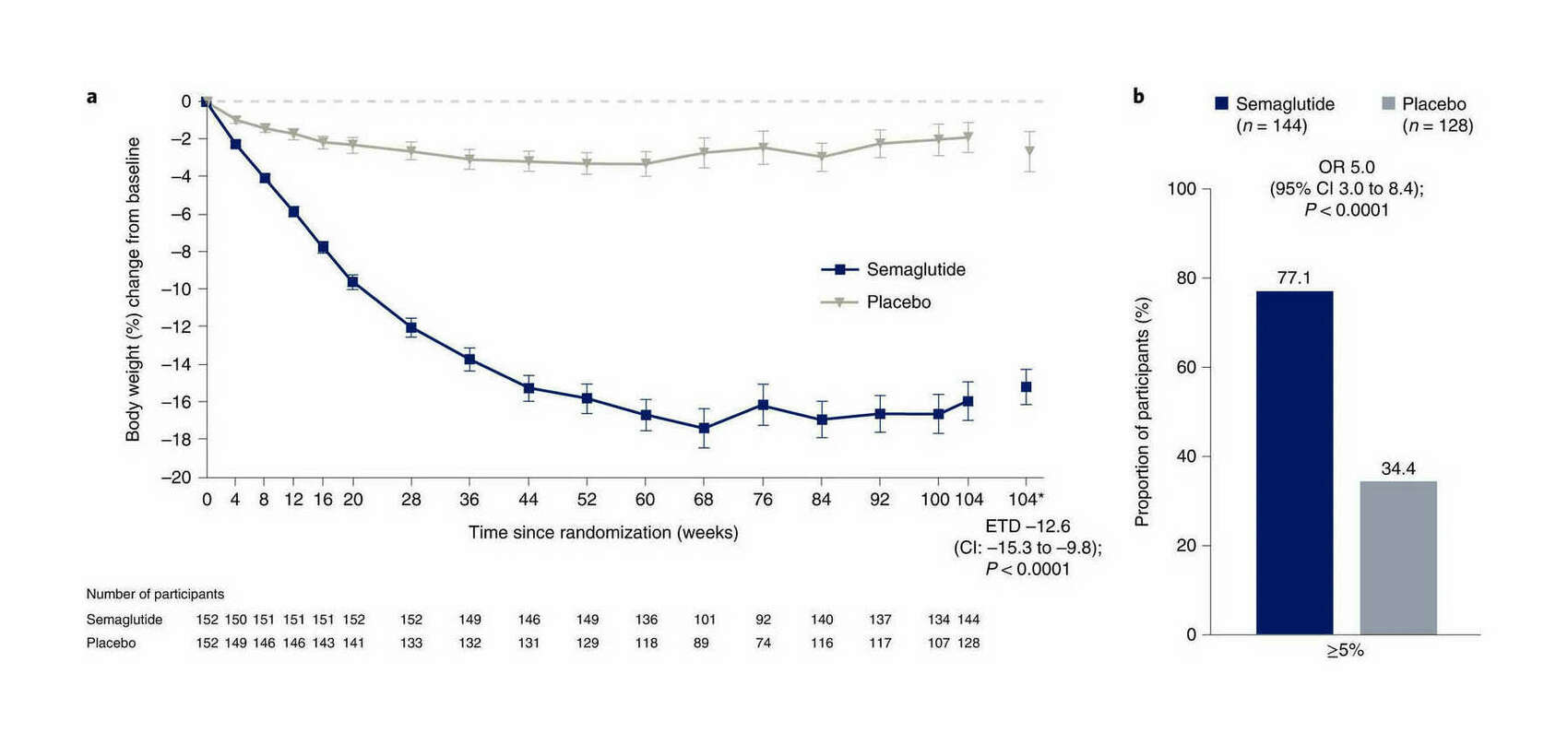
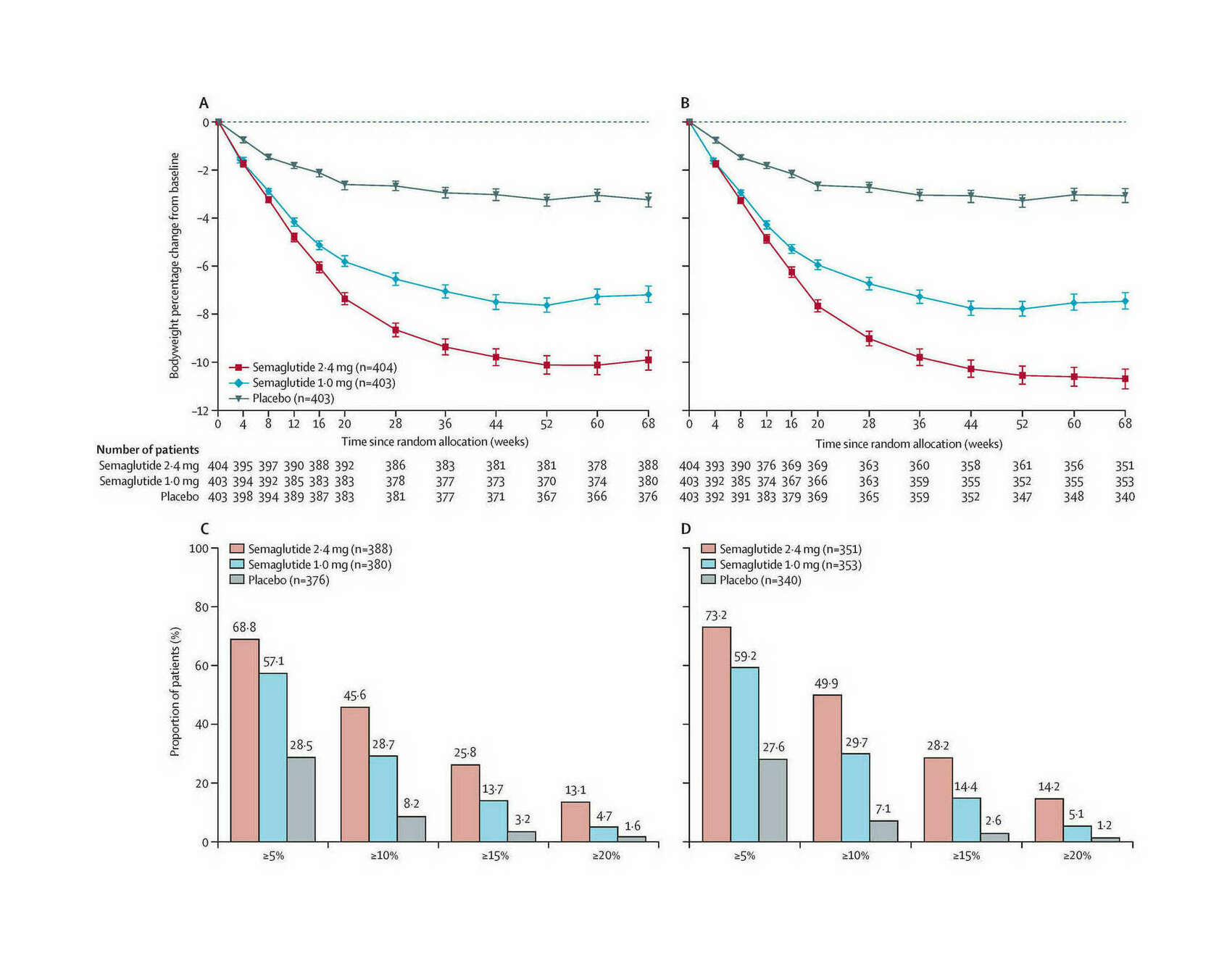
/doc/longevity/glp/semaglutide/2021-wilding-figure-ad-weightlosseffects.png: -
https://chadnauseam.com/random/semaglutide-has-changed-the-world/: -
https://trevorklee.com/should-you-take-metformin-for-longevity/: -
https://trevorklee.substack.com/p/obesitys-relationship-with-type-2 -
https://trevorklee.substack.com/p/pharmacokinetics-drug-developments#footnote-4-137517922 -
https://twitter.com/CharlesCMann/status/1673310755079192579: -
https://twitter.com/DanielJDrucker/status/1591171488002232320: -
https://twitter.com/RosieCampbell/status/1636050117202694144: -
https://www.astralcodexten.com/p/highlights-from-the-comments-on-semaglutide -
https://www.nytimes.com/2023/05/16/well/live/ozempic-alternatives-semaglutide.html -
https://www.nytimes.com/2023/11/03/well/mind/ozempic-weight-loss-antidepressants-antipsychotics.html -
https://www.overcomingbias.com/p/one-pill-to-break-us-allhtml -
https://www.science.org/content/blog-post/ozempic-and-other-glp-1-drugs-more-people-realize
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Link Bibliography
-
https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2816824: “Estimated Sustainable Cost-Based Prices for Diabetes Medicines”, Melissa J. Barber, Dzintars Gotham, Helen Bygrave, Christa Cepuch -
https://www.nature.com/articles/s42255-023-00943-3: “Randomized Open-Label Trial of Semaglutide and Dapagliflozin in Patients With Type 2 Diabetes of Different Pathophysiology”, Chinmay Dwibedi, Ola Ekström, Jasmine Brandt, Martin Adiels, Stefan Franzén, Birgitta Abrahamsson, Anders H. Rosengren -
2023-richards.pdf: “Substantial Decrease in Alcohol Use Disorder Symptoms Secondary to Semaglutide Therapy for Weight Loss: A Case Series”, Jesse R. Richards, Madisen Fae Dorand, Kyleigh Royal, Lana Mnajjed, Maria Paszkowiak, W. Kyle Simmons -
2023-lincoff.pdf: “Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes”, -
2023-lingvay.pdf: “A Revolution in Obesity Treatment”, Ildiko Lingvay, Shubham Agarwal -
https://www.wsj.com/health/healthcare/ozempic-mounjaro-weight-loss-drug-cost-32fc3555: “To Pay for Weight Loss Drugs, Some Take Second Jobs, Ring Up Credit Card Debts: Some People Pay More Than $10,000 a Year Out-Of-Pocket for Ozempic and Mounjaro”, Stephanie Armour -
https://www.wired.com/story/obesity-drugs-researcher-interview-ozempic-wegovy/: “What the Scientists Who Pioneered Weight-Loss Drugs Want You to Know”, Matt Reynolds -
https://www.theatlantic.com/health/archive/2023/05/ozempic-addictive-behavior-drinking-smoking/674098/: “Ozempic’s Next Act: People Taking the Drug for Weight Loss Say They Have Also Stopped Drinking, Smoking, Shopping, and Even Nail-Biting”, Sarah Zhang -
https://www.sciencedirect.com/science/article/pii/S2212877822001028: “Next Generation GLP-1/GIP/glucagon Triple Agonists Normalize Body Weight in Obese Mice”, -
https://link.springer.com/article/10.1007/s40262-021-01025-x: “Clinical Pharmacokinetics of Oral Semaglutide: Analyses of Data from Clinical Pharmacology Trials”, Rune V. Overgaard, Andrea Navarria, Steen H. Ingwersen, Tine A. Bækdal, Rasmus Juul Kildemoes -
2021-lundgren.pdf: “Healthy Weight Loss Maintenance With Exercise, Liraglutide, or Both Combined”, -
https://www.sciencedirect.com/science/article/pii/S2212877821000156: “The Gut-Brain Axis: Identifying New Therapeutic Approaches for Type 2 Diabetes, Obesity, and Related Disorders”, Paul Richards, Nancy A. Thornberry, Shirly Pinto -
2020-kushner.pdf: “Semaglutide 2.4 Mg for the Treatment of Obesity: Key Elements of the STEP Trials 1 to 5”, -
https://www.sciencedirect.com/science/article/pii/S0028390819300541: “Glucagon-Like Peptide-1 Receptors within the Nucleus of the Solitary Tract Regulate Alcohol-Mediated Behaviors in Rodents”, Daniel Vallöf, Jesper Vestlund, Elisabet Jerlhag -
https://www.frontiersin.org/articles/10.3389/fendo.2019.00155/full: “The Discovery and Development of Liraglutide and Semaglutide”, Lotte Bjerre Knudsen, Jesper Lau -
2017-davies.pdf: “Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial”, Melanie Davies, Thomas R. Pieber, Marie-Louise Hartoft-Nielsen, Oluf K. H. Hansen, Serge Jabbour, Julio Rosenstock