- See Also
-
Links
- “De Novo Variants in the Non-Coding Spliceosomal SnRNA Gene RNU4-2 Are a Frequent Cause of Syndromic Neurodevelopmental Disorders”, Chen et al 2024
- “Widespread Recessive Effects on Common Diseases in a Cohort of 44,000 British Pakistanis and Bangladeshis With High Autozygosity”, Heng et al 2024
- “Dissecting the Contribution of Common Variants to Risk of Rare Neurodevelopmental Conditions”, Huang et al 2024
- “Narrowing the Diagnostic Gap: Genomes, Episignatures, Long-Read Sequencing, and Health Economic Analyses in an Exome-Negative Intellectual Disability Cohort”, Dias et al 2024
- “Imputation of Structural Variants Using a Multi-Ancestry Long-Read Sequencing Panel Enables Identification of Disease Associations”, Noyvert et al 2023
- “Whole-Genome Sequencing of Half-A-Million UK Biobank Participants”, Consortium et al 2023
- “Human Deleterious Mutation Rate Implies High Fitness Variance, With Declining Mean Fitness Compensated by Rarer Beneficial Mutations of Larger Effect”, Matheson et al 2023
- “From Target Discovery to Clinical Drug Development With Human Genetics”, Trajanoska et al 2023
- “Rare Coding Variants in Schizophrenia-Associated Genes Affect Generalised Cognition in the UK Biobank”, Fenner et al 2023
- “Somatic Mutations in Human Ageing: New Insights from DNA Sequencing and Inherited Mutations”, Chatsirisupachai & Magalhães 2023
- “The Vanishing Family: They All Have a 50-50 Chance of Inheriting a Cruel Genetic Mutation—Which Means Disappearing into Dementia in Middle Age. This Is the Story of What It’s like to Live With Those Odds”, Kolker 2023
- “Phenotypic Effects of Genetic Variants Associated With Autism”, Rolland et al 2023
- “South Asian Medical Cohorts Reveal Strong Founder Effects and High Rates of Homozygosity”, Wall et al 2023
- “Estimating the Parental Age Effect on Intelligence With Controlling for Confounding Effects from Genotypic Differences”, Wang 2023
- “Molecular Basis of FAAH-OUT-Associated Human Pain Insensitivity”, Mikaeili et al 2023
- “DNA Repair and Anti-Cancer Mechanisms in the Longest-Living Mammal: the Bowhead Whale”, Firsanov et al 2023
- “Predicting ExWAS Findings from GWAS Data: a Shorter Path to Causal Genes”, Liang et al 2023b
- “Common and Rare Variant Associations With Latent Traits Underlying Depression, Bipolar Disorder, and Schizophrenia”, Dattani et al 2023
- “Schizophrenia-Associated Somatic Copy-Number Variants from 12,834 Cases Reveal Recurrent NRXN1 and ABCB11 Disruptions”, Maury et al 2023
- “Large-Scale Exome Sequence Analysis Identifies Sex- and Age-Specific Determinants of Obesity”, Kaisinger et al 2023
- “Quantifying Constraint in Human Mitochondrial DNA”, Lake et al 2022
- “Heritability of de Novo Germline Mutation Reveals a Contribution from Paternal but Not Maternal Genetic Factors”, Hwang et al 2022
- “Cell Tree Rings: the Shape of Somatic Evolution As a Human Aging Timer”, Csordas et al 2022
- “Rare and Common Genetic Determinants of Metabolic Individuality and Their Effects on Human Health”, Surendran et al 2022
- “Accurate Detection of Shared Genetic Architecture from GWAS Summary Statistics in the Small-Sample Context”, Willis & Wallace 2022
- “Declining Autozygosity over Time: an Exploration in over 1 Million Individuals from Three Diverse Cohorts”, Colbert et al 2022
- “Influences of Rare Protein-Coding Genetic Variants on the Human Plasma Proteome in 50,829 UK Biobank Participants”, Dhindsa et al 2022
- “Developmental Implications of Genetic Testing for Physical Indications”, Baribeau et al 2022
- “Genome-Wide Prediction of Disease Variants With a Deep Protein Language Model”, Brandes et al 2022
- “Genomic Health Is Dependent on Population Demographic History”, Wootton & Shafer 2022
- “Nationwide Genomic Biobank in Mexico Unravels Demographic History and Complex Trait Architecture from 6,057 Individuals”, Sohail et al 2022
- “Polygenic Architecture of Rare Coding Variation across 400,000 Exomes”, Weiner et al 2022
- “Complex Traits and Candidate Genes: Estimation of Genetic Variance Components Across Modes of Inheritance”, Feldmann et al 2022
- “The Impact of Rare Protein Coding Genetic Variation on Adult Cognitive Function”, Chen et al 2022
- “Rare Genetic Variants Impact Muscle Strength”, Huang et al 2022
- “Genetic Prevalence and Clinical Relevance of Canine Mendelian Disease Variants in over One Million Dogs”, Donner et al 2022
- “The Female Protective Effect against Autism Spectrum Disorder”, Wigdor et al 2022
- “Using Genomics to Fight Extinction: Quantifying Fitness of Wild Organisms from Genomic Data Alone Is a Challenging Frontier”, Grueber & Sunnucks 2022
- “The Critically Endangered Vaquita Is Not Doomed to Extinction by Inbreeding Depression”, Robinson et al 2022
- “Polygenic Risk Score As a Possible Tool for Identifying Familial Monogenic Causes of Complex Diseases”, Lu et al 2022
- “The Contributions of Rare Inherited and Polygenic Risk to ASD in Multiplex Families”, Chang et al 2022
- “Exome-Wide Screening Identifies Novel Rare Risk Variants for Major Depression Disorder”, Cheng et al 2022b
- “Integrating Whole-Genome Sequencing With Multi-Omic Data Reveals the Impact of Structural Variants on Gene Regulation in the Human Brain”, Vialle et al 2022
- “Characterization of Arabian Peninsula Whole Exomes: Exploring High Inbreeding Features”, Ferreira et al 2022
- “Genome-Wide Analyses of ADHD Identify 27 Risk Loci, Refine the Genetic Architecture and Implicate Several Cognitive Domains”, Demontis et al 2022
- “Genetic Risk Factors Have a Substantial Impact on Healthy Life Years”, Jukarainen et al 2022
- “From Variant to Function in Human Disease Genetics”, Lappalainen & MacArthur 2022
- “Life Histories of Myeloproliferative Neoplasms Inferred from Phylogenies”, Williams et al 2022
- “Ultra-Rapid Nanopore Genome Sequencing in a Critical Care Setting”, Gorzynski et al 2022
- “Rare Genetic Variants Correlate With Better Processing Speed”, Song et al 2022
- “Rare Schizophrenia Risk Variant Burden Is Conserved in Diverse Human Populations”, Liu et al 2022
- “Schizophrenia-Associated Somatic Copy Number Variants from 12,834 Cases Reveal Contribution to Risk and Recurrent, Isoform-Specific NRXN1 Disruptions”, Maury et al 2022
- “Rare Genetic Variants in Genes and Loci Linked to Dominant Monogenic Developmental Disorders Cause Milder Related Phenotypes in the General Population”, Kingdom et al 2022
- “Dominant Cone Rod Dystrophy, Previously Assigned to a Missense Variant in RIMS1, Is Fully Explained by Co-Inheritance of a Dominant Allele of PROM1”, Martin-Gutierrez et al 2022
- “The Origins and Functional Effects of Postzygotic Mutations throughout the Human Lifespan”, Rockweiler et al 2021
- “High-Impact Rare Genetic Variants in Severe Schizophrenia”, Zoghbi et al 2021
- “Familial Risk and Heritability of Intellectual Disability: a Population-Based Cohort Study in Sweden”, Lichtenstein et al 2021
- “CONGA: Copy Number Variation Genotyping in Ancient Genomes and Low-Coverage Sequencing Data”, Soylev et al 2021
- “A Spectrum of Recessiveness among Mendelian Disease Variants in UK Biobank”, Barton et al 2021
- “Fine-Scale Population Structure and Demographic History of British Pakistanis”, Arciero et al 2021
- “The Effect of Inbreeding, Body Size and Morphology on Health in Dog Breeds”, Bannasch et al 2021
- “Deletion of Loss-Of-Function-Intolerant Genes and Risk of 5 Psychiatric Disorders”, Wainberg et al 2021
- “Exploring the Relationships between Autozygosity, Educational Attainment, and Cognitive Ability in a Contemporary, Trans-Ancestral American Sample”, Colbert et al 2021
- “Deep Learning Enables Genetic Analysis of the Human Thoracic Aorta”, Pirruccello et al 2021
- “Comparing Copy Number Variations in a Danish Case Cohort of Individuals With Psychiatric Disorders”, Sánchez et al 2021
- “The Sequences of 150,119 Genomes in the UK Biobank”, Halldorsson et al 2021
- “The Impact of Rare Germline Variants on Human Somatic Mutation Processes”, Vali-Pour et al 2021
- “100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report”, Investigators 2021
- “Influences of Rare Copy Number Variation on Human Complex Traits”, Hujoel et al 2021
- “Rare Variant Aggregation in 148,508 Exomes Identifies Genes Associated With Proxy Alzheimer’s Disease”, Wightman et al 2021
- “Integrating de Novo and Inherited Variants in over 42,607 Autism Cases Identifies Mutations in New Moderate Risk Genes”, Zhou et al 2021
- “How Rare and Common Risk Variation Jointly Affect Liability for Autism Spectrum Disorder”, Klei et al 2021
- “A General Framework for Identifying Rare Variant Combinations in Complex Disorders”, Pounraja & Girirajan 2021
- “Extreme Purifying Selection against Point Mutations in the Human Genome”, Dukler et al 2021
- “Rates of Contributory de Novo Mutation in High and Low-Risk Autism Families”, Yoon et al 2021
- “Partitioning Gene-Level Contributions to Complex-Trait Heritability by Allele Frequency Identifies Disease-Relevant Genes”, Burch et al 2021
- “Differences in the Genetic Architecture of Common and Rare Variants in Childhood, Persistent and Late-Diagnosed Attention Deficit Hyperactivity Disorder”, Rajagopal et al 2021
- “Genetic Correlates of Phenotypic Heterogeneity in Autism”, Warrier et al 2021
- “Sequencing of 640,000 Exomes Identifies GPR75 Variants Associated With Protection from Obesity”, Akbari et al 2021
- “Finding Genes That Control Body Weight: DNA Exome Sequencing at Scale Reveals Unknown Human Biology of Adiposity”, Yeo & O’Rahilly 2021
- “Genomic Partitioning of Inbreeding Depression in Humans”, Yengo et al 2021
- “Exome Sequencing in Obsessive-Compulsive Disorder Reveals a Burden of Rare Damaging Coding Variants”, Halvorsen et al 2021
- “Recovery of Trait Heritability from Whole Genome Sequence Data”, Wainschtein et al 2021
- “Ultra-Rare, Rare, and Common Genetic Variant Analysis Converge to Implicate Negative Selection and Neuronal Processes in the Aetiology of Schizophrenia”, Akingbuwa et al 2021
- “Lack of Transgenerational Effects of Ionizing Radiation Exposure from the Chernobyl Accident”, Yeager et al 2021
- “A Phenotypic Spectrum of Autism Is Attributable to the Combined Effects of Rare Variants, Polygenic Risk and Sex”, Antaki et al 2021
- “Structural Variants in Chinese Population and Their Impact on Phenotypes, Diseases and Population Adaptation”, Wu et al 2021
- “Polygenic Burden Has Broader Impact on Health, Cognition, and Socioeconomic Outcomes Than Most Rare and High-Risk Copy Number Variants”, Saarentaus et al 2021
- “A Cross-Disorder Dosage Sensitivity Map of the Human Genome”, Collins et al 2021
- “Protein-Coding Repeat Polymorphisms Strongly Shape Diverse Human Phenotypes”, Mukamel et al 2021
- “The Landscape of Somatic Mutation in Cerebral Cortex of Autistic and Neurotypical Individuals Revealed by Ultra-Deep Whole-Genome Sequencing”, Rodin et al 2021
- “Large Mosaic Copy Number Variations Confer Autism Risk”, Sherman et al 2021b
- “Long Tracks of Homozygosity Predict the Severity of Alcohol Use Disorders in an American Indian Population”, Peng & Ehlers 2021
- “Exome Sequencing and Analysis of 454,787 UK Biobank Participants”, Backman et al 2021
- “Long Read Sequencing of 3,622 Icelanders Provides Insight into the Role of Structural Variants in Human Diseases and Other Traits”, Beyter et al 2020
- “A Broad Exome Study of the Genetic Architecture of Asthma Reveals Novel Patient Subgroups”, Cameron-Christie et al 2020
- “Rare Genetic Variation Underlying Human Diseases and Traits: Results from 200,000 Individuals in the UK Biobank”, Jurgens et al 2020
- “Discovery of Rare Variants Associated With Blood Pressure Regulation through Meta-Analysis of 1.3 Million Individuals”, Surendran et al 2020
- “Exome Sequencing and Characterization of 49,960 Individuals in the UK Biobank”, Hout et al 2020
- “Mutations in Metabotropic Glutamate Receptor 1 Contribute to Natural Short Sleep Trait”, Shi et al 2020
- “Exome Sequencing Identifies Rare Coding Variants in 10 Genes Which Confer Substantial Risk for Schizophrenia”, Singh et al 2020
- “Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia”, Consortium et al 2020
- “Effect Sizes of Deletions and Duplications on Autism Risk Across the Genome”, Douard et al 2020
- “Novel Ultra-Rare Exonic Variants Identified in a Founder Population Implicate Cadherins in Schizophrenia”, Lencz et al 2020
- “Reconstructing the History of Founder Events Using Genome-Wide Patterns of Allele Sharing across Individuals”, Tournebize et al 2020
- “Whole-Exome Imputation within UK Biobank Powers Rare Coding Variant Association and Fine-Mapping Analyses”, Barton et al 2020
- “Exome-Wide Association Studies in General and Long-Lived Populations Identify Genetic Variants Related to Human Age”, Sin-Chan et al 2020
- “Genomic Analyses Implicate Noncoding de Novo Variants in Congenital Heart Disease”, Richter et al 2020
- “Genetic Ancestry Analysis on >93,000 Individuals Undergoing Expanded Carrier Screening Reveals Limitations of Ethnicity-Based Medical Guidelines”, Kaseniit et al 2020
- “The Burden of Rare Protein-Truncating Genetic Variants on Human Lifespan”, Liu et al 2020
- “An Integrated Polygenic and Clinical Risk Tool Enhances Coronary Artery Disease Prediction”, Aguilera et al 2020
- “Sex-Biased Reduction in Reproductive Success Drives Selective Constraint on Human Genes”, Gardner et al 2020
- “Etiology of Autism Spectrum Disorders and Autistic Traits Over Time”, Taylor et al 2020
- “Estimating the Effect-Size of Gene Dosage on Cognitive Ability across the Coding Genome”, Huguet et al 2020
- “Inbreeding and Inbreeding Depression in Linebred Beef Cattle”, Hieber 2020
- “Whole-Genome Sequencing of Rare Disease Patients in a National Healthcare System”, Ouwehand et al 2020
- “Figure 6. The Contributions of Ultra-Rare PTVs [protein-Truncating Variants] to Schizophrenia Risk. A: Genetic Architecture of Schizophrenia. Statistically-Significant Genetic Associations for Schizophrenia from the Most Recent GWAS, CNV, and Sequencing Studies Are Displayed. The In-Sample Odds Ratio Is Plotted against the Minor Allele Frequency in the General Population. The Color of Each Dot Corresponds to the Source of the Association, and the Size of the Dot to the Odds Ratio. The Shaded Area Represented the LOESS-Smoothed Lines of the Upper and Lower Bounds of the Point Estimates...Because Schizophrenia As a Trait Is under Strong Selection^38–40^, We Expect That URVs of Large Effect to Be Frequently de Novo or of Very Recent Origin and Contribute to Risk in Only a Fraction of Diagnosed Patients.”
- “Extended Data Figure 2: GWAS Progress over Time. The Relationship of GWAS Associations to Sample-Size Is Shown in This Plot With Selected SCZ GWAS Meta-Analyses of the past 11 Years. The X-Axis Shows Number of Cases. The Y-Axis Shows the Number of Independent Loci Discovered With at Least One Genome-Wide Statistically-Significant Index SNP in the Discovery Meta-Analysis (eg. without Replication Data)...The Slope of ~4 Newly Discovered Loci per 1000 Cases 2013–2019 Increased to a Slope of ~6 With the Latest Sample-Size Increase.”
- “Rare Genetic Variants Associated With Sudden Cardiac Death in Adults”, Khera et al 2019
- “Mutant Neuropeptide S Receptor Reduces Sleep Duration With Preserved Memory Consolidation”, Xing et al 2019
- “Germline Burden of Rare Damaging Variants Negatively Affects Human Healthspan and Lifespan”, Shindyapina et al 2019
- “Extreme Inbreeding in a European Ancestry Sample from the Contemporary UK Population”, Yengo et al 2019
- “Insights about Variation in Meiosis from 31,228 Human Sperm Genomes”, Bell et al 2019
- “The Family That Feels Almost No Pain: An Italian Clan’s Curious Insensitivity to Pain Has Piqued the Interest of Geneticists Seeking a New Understanding of How to Treat Physical Suffering”, Shaer 2019
- “Symposium Review: The Genomic Architecture of Inbreeding: How Homozygosity Affects Health and Performance”, Baes et al 2019
- “Crowdfunded Whole-Genome Sequencing of the Celebrity Cat Lil BUB Identifies Causal Mutations for Her Osteopetrosis and Polydactyly”, Bridavsky et al 2019
- “Microdeletion in a FAAH Pseudogene Identified in a Patient With High Anandamide Concentrations and Pain Insensitivity”, Habib et al 2019
- “Whole-Genome Deep-Learning Analysis Identifies Contribution of Noncoding Mutations to Autism Risk”, Zhou et al 2019b
- “The Genetic Basis of Inbreeding Depression in Potato”, Zhang et al 2019
- “A Rare Mutation of Β1-Adrenergic Receptor Affects Sleep/Wake Behaviors”, Shi et al 2019b
- “Phenome-Wide Burden of Copy-Number Variation in the UK Biobank”, Aguirre et al 2019
- “The Human-Specific BOLA2 Duplication Modifies Iron Homeostasis and Anemia Predisposition in Chromosome 16p11.2 Autism Individuals”, Giannuzzi et al 2019
- “Schizophrenia Risk Conferred by Protein-Coding de Novo Mutations”, Howrigan et al 2018
- “Pfizer Terminates Domagrozumab (PF-06252616) Clinical Studies for the Treatment of Duchenne Muscular Dystrophy”, Pfizer 2018
- “Quantifying the Effects of 16p11.2 Copy Number Variants on Brain Structure: A Multisite Genetic-First Study”, Martin-Brevet et al 2018
- “LY2495655, an Antimyostatin Antibody, in Pancreatic Cancer: a Randomized, Phase 2 Trial”, Golan et al 2018
- “Common Genetic Variants Contribute to Risk of Rare Severe Neurodevelopmental Disorders”, Niemi et al 2018
- “Analysis of the Genetic Basis of Height in Large Jewish Nuclear Families”, Zeevi et al 2018
- “Frequency and Distribution of 152 Genetic Disease Variants in over 100,000 Mixed Breed and Purebred Dogs”, Donner et al 2018
- “Relationships between Estimated Autozygosity and Complex Traits in the UK Biobank”, Johnson et al 2018
- “Myostatin: 20 Years Later”, Kostyunina et al 2018
- “An Analytical Framework for Whole-Genome Sequence Association Studies and Its Implications for Autism Spectrum Disorder”, Werling et al 2018
- “1 in 38 Individuals at Risk of a Dominant Medically Actionable Disease”, Haer-Wigman et al 2018
- “Secondary Findings from Clinical Genomic Sequencing: Prevalence, Patient Perspectives, Family History Assessment, and Health-Care Costs from a Multisite Study”, Hart et al 2018
- “Medical Consequences of Pathogenic CNVs in Adults: Analysis of the UK Biobank”, Crawford et al 2018
- “Measuring and Estimating the Effect Sizes of Copy Number Variants on General Intelligence in Community-Based Samples”, Huguet et al 2018
- “Singleton Variants Dominate the Genetic Architecture of Human Gene Expression”, Hernandez et al 2017
- “CNV-Association Meta-Analysis in 191,161 European Adults Reveals New Loci Associated With Anthropometric Traits”, Macé et al 2017
- “Quantification of Frequency-Dependent Genetic Architectures and Action of Negative Selection in 25 UK Biobank Traits”, Schoech et al 2017
- “A Genome-Wide Association Study for Extremely High Intelligence”, Zabaneh et al 2017
- “The Surprising Implications of Familial Association in Disease Risk”, Valberg et al 2017
- “Quantifying the Impact of Rare and Ultra-Rare Coding Variation across the Phenotypic Spectrum”, Ganna et al 2017
- “Genomic Analysis of Family Data Reveals Additional Genetic Effects on Intelligence and Personality”, Hill et al 2017
- “Prevalence and Architecture of de Novo Mutations in Developmental Disorders”, McRae et al 2017
- “Excess of Genomic Defects in a Woolly Mammoth on Wrangel Island”, Rogers & Slatkin 2017
- “Parental Influence on Human Germline de Novo Mutations in 1,548 Trios from Iceland”, Jónsson et al 2017
- “Inequality in Genetic Cancer Risk Suggests Bad Genes rather than Bad Luck”, Stensrud & Valberg 2017
- “Polygenic Transmission Disequilibrium Confirms That Common and Rare Variation Act Additively to Create Risk for Autism Spectrum Disorders”, Weiner et al 2016
- “Trans-Ancestry Meta-Analyses Identify Rare and Common Variants Associated With Blood Pressure and Hypertension”, Surendran et al 2016
- “Clinical Utility of Expanded Carrier Screening: Reproductive Behaviors of At-Risk Couples”, Ghiossi et al 2016
- “Extreme Distribution of Deleterious Variation in a Historically Small and Isolated Population—Insights from the Greenlandic Inuit”, Pedersen et al 2016
- “A Prospective Study of Sudden Cardiac Death among Children and Young Adults”, Bagnall et al 2016
- “Cognitive Performance Among Carriers of Pathogenic Copy Number Variants: Analysis of 152,000 UK Biobank Subjects”, Kendall et al 2016
- “Family-Specific Variants and the Limits of Human Genetics”, Shirts et al 2016
- “Whole-Genome Sequencing of Quartet Families With Autism Spectrum Disorder”
- “The Relative Contribution of Common and Rare Genetic Variants to ADHD”, Martin 2015
- “Complete Genomes Reveal Signatures of Demographic and Genetic Declines in the Woolly Mammoth”, Palkopoulou et al 2015
- “Directional Dominance on Stature and Cognition in Diverse Human Populations”, Joshi et al 2015
- “Synaptic, Transcriptional and Chromatin Genes Disrupted in Autism”, Rubeis et al 2014
- “Estimating the Inbreeding Depression on Cognitive Behavior: A Population Based Study of Child Cohort”, Fareed & Afzal 2014
- “A Novel BHLHE41 Variant Is Associated With Short Sleep and Resistance to Sleep Deprivation in Humans”, Pellegrino et al 2014
- “Large-Scale Genomics Unveils the Genetic Architecture of Psychiatric Disorders”, Gratten et al 2014
- “The Contribution of de Novo Coding Mutations to Autism Spectrum Disorder”, Iossifov et al 2014
- “The Effect of Paternal Age on Offspring Intelligence and Personality When Controlling for Paternal Trait Level”, Arslan et al 2013
- “Analysis of 6,515 Exomes Reveals the Recent Origin of Most Human Protein-Coding Variants”, Fu et al 2013
- “The Incidence of Leukemia, Lymphoma and Multiple Myeloma among Atomic Bomb Survivors: 1950-2001”, Hsu et al 2013
- “Range of Genetic Mutations Associated With Severe Non-Syndromic Sporadic Intellectual Disability: an Exome Sequencing Study”, Rauch et al 2012
- “CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics”, Malhotra & Sebat 2012
- “Heritability of Performance Deficit Accumulation during Acute Sleep Deprivation in Twins”, Kuna et al 2012
- “Common Variants Show Predicted Polygenic Effects on Height in the Tails of the Distribution, Except in Extremely Short Individuals”, Chan et al 2011
- “Rare Copy Number Deletions Predict Individual Variation in Intelligence”, Yeo et al 2010
- “Genomic Insights into Early-Onset Obesity”, Choquet & Meyre 2010
- “Population-Based Carrier Screening for Cystic Fibrosis in Victoria: The First 3 Years Experience”, Massie et al 2009
- “An Expressed fgf4 Retrogene Is Associated With Breed-Defining Chondrodysplasia in Domestic Dogs.”, Parker et al 2009
- “The Transcriptional Repressor DEC2 Regulates Sleep Length in Mammals”, He et al 2009
- “The VNTR 2 Repeat in MAOA and Delinquent Behavior in Adolescence and Young Adulthood: Associations and MAOA Promoter Activity”, Guo et al 2008
- “Language and Communicative Development in Williams Syndrome”, Mervis & Becerra 2007
- “Genetic Enhancement of Cognition in a Kindred With Cone-Rod Dystrophy due to RIMS1 Mutation”, Sisodiya et al 2007
- “Self-Management of Fatal Familial Insomnia. Part 2: Case Report”, Schenkein & Montagna 2006
- “Myostatin Mutation Associated With Gross Muscle Hypertrophy in a Child”, Schuelke et al 2004
- “Influence of Five Years of Antenatal Screening on the Paediatric Cystic Fibrosis Population in One Region”, Cunningham & Marshall 1998
- “When Kim Goodsell Discovered That She Had Two Extremely Rare Genetic Diseases, She Taught Herself Genetics to Help Find out Why.”
- “Natural History of Ashkenazi Intelligence”
- “The Sports Gene: Inside the Science of Extraordinary Athletic Performance”
- “Fathers Bequeath More Mutations As They Age: Genome Study May Explain Links between Paternal Age and Conditions such as Autism”
- “Monkeys Genetically Modified to Show Autism Symptoms: But It Is Unclear How Well the Results Match the Condition in Humans”
- “A Genome-Wide Analysis of Putative Functional and Exonic Variation Associated With Extremely High Intelligence”
- “A Gene That Makes You Need Less Sleep?”
- “What's Behind Many Mystery Ailments? Genetic Mutations, Study Finds”
- “Thinking Positively: The Genetics of High Intelligence”
- “Why Do Humans Still Have a Gene That Increases the Risk of Alzheimer’s?”
- Sort By Magic
- Wikipedia
- Miscellaneous
- Link Bibliography
See Also
Links
“De Novo Variants in the Non-Coding Spliceosomal SnRNA Gene RNU4-2 Are a Frequent Cause of Syndromic Neurodevelopmental Disorders”, Chen et al 2024
“Widespread Recessive Effects on Common Diseases in a Cohort of 44,000 British Pakistanis and Bangladeshis With High Autozygosity”, Heng et al 2024
“Dissecting the Contribution of Common Variants to Risk of Rare Neurodevelopmental Conditions”, Huang et al 2024
Dissecting the contribution of common variants to risk of rare neurodevelopmental conditions
“Narrowing the Diagnostic Gap: Genomes, Episignatures, Long-Read Sequencing, and Health Economic Analyses in an Exome-Negative Intellectual Disability Cohort”, Dias et al 2024
“Imputation of Structural Variants Using a Multi-Ancestry Long-Read Sequencing Panel Enables Identification of Disease Associations”, Noyvert et al 2023
“Whole-Genome Sequencing of Half-A-Million UK Biobank Participants”, Consortium et al 2023
Whole-genome sequencing of half-a-million UK Biobank participants
“Human Deleterious Mutation Rate Implies High Fitness Variance, With Declining Mean Fitness Compensated by Rarer Beneficial Mutations of Larger Effect”, Matheson et al 2023
“From Target Discovery to Clinical Drug Development With Human Genetics”, Trajanoska et al 2023
From target discovery to clinical drug development with human genetics
“Rare Coding Variants in Schizophrenia-Associated Genes Affect Generalised Cognition in the UK Biobank”, Fenner et al 2023
“Somatic Mutations in Human Ageing: New Insights from DNA Sequencing and Inherited Mutations”, Chatsirisupachai & Magalhães 2023
Somatic mutations in human ageing: New insights from DNA sequencing and inherited mutations
“The Vanishing Family: They All Have a 50-50 Chance of Inheriting a Cruel Genetic Mutation—Which Means Disappearing into Dementia in Middle Age. This Is the Story of What It’s like to Live With Those Odds”, Kolker 2023
“Phenotypic Effects of Genetic Variants Associated With Autism”, Rolland et al 2023
Phenotypic effects of genetic variants associated with autism
“South Asian Medical Cohorts Reveal Strong Founder Effects and High Rates of Homozygosity”, Wall et al 2023
South Asian medical cohorts reveal strong founder effects and high rates of homozygosity
“Estimating the Parental Age Effect on Intelligence With Controlling for Confounding Effects from Genotypic Differences”, Wang 2023
“Molecular Basis of FAAH-OUT-Associated Human Pain Insensitivity”, Mikaeili et al 2023
Molecular basis of FAAH-OUT-associated human pain insensitivity
“DNA Repair and Anti-Cancer Mechanisms in the Longest-Living Mammal: the Bowhead Whale”, Firsanov et al 2023
DNA repair and anti-cancer mechanisms in the longest-living mammal: the bowhead whale
“Predicting ExWAS Findings from GWAS Data: a Shorter Path to Causal Genes”, Liang et al 2023b
Predicting ExWAS findings from GWAS data: a shorter path to causal genes
“Common and Rare Variant Associations With Latent Traits Underlying Depression, Bipolar Disorder, and Schizophrenia”, Dattani et al 2023
“Schizophrenia-Associated Somatic Copy-Number Variants from 12,834 Cases Reveal Recurrent NRXN1 and ABCB11 Disruptions”, Maury et al 2023
“Large-Scale Exome Sequence Analysis Identifies Sex- and Age-Specific Determinants of Obesity”, Kaisinger et al 2023
Large-scale exome sequence analysis identifies sex- and age-specific determinants of obesity
“Quantifying Constraint in Human Mitochondrial DNA”, Lake et al 2022
“Heritability of de Novo Germline Mutation Reveals a Contribution from Paternal but Not Maternal Genetic Factors”, Hwang et al 2022
“Cell Tree Rings: the Shape of Somatic Evolution As a Human Aging Timer”, Csordas et al 2022
Cell Tree Rings: the shape of somatic evolution as a human aging timer
“Rare and Common Genetic Determinants of Metabolic Individuality and Their Effects on Human Health”, Surendran et al 2022
Rare and common genetic determinants of metabolic individuality and their effects on human health
“Accurate Detection of Shared Genetic Architecture from GWAS Summary Statistics in the Small-Sample Context”, Willis & Wallace 2022
“Declining Autozygosity over Time: an Exploration in over 1 Million Individuals from Three Diverse Cohorts”, Colbert et al 2022
“Influences of Rare Protein-Coding Genetic Variants on the Human Plasma Proteome in 50,829 UK Biobank Participants”, Dhindsa et al 2022
“Developmental Implications of Genetic Testing for Physical Indications”, Baribeau et al 2022
Developmental implications of genetic testing for physical indications
“Genome-Wide Prediction of Disease Variants With a Deep Protein Language Model”, Brandes et al 2022
Genome-wide prediction of disease variants with a deep protein language model
“Genomic Health Is Dependent on Population Demographic History”, Wootton & Shafer 2022
Genomic health is dependent on population demographic history
“Nationwide Genomic Biobank in Mexico Unravels Demographic History and Complex Trait Architecture from 6,057 Individuals”, Sohail et al 2022
“Polygenic Architecture of Rare Coding Variation across 400,000 Exomes”, Weiner et al 2022
Polygenic architecture of rare coding variation across 400,000 exomes
“Complex Traits and Candidate Genes: Estimation of Genetic Variance Components Across Modes of Inheritance”, Feldmann et al 2022
“The Impact of Rare Protein Coding Genetic Variation on Adult Cognitive Function”, Chen et al 2022
The impact of rare protein coding genetic variation on adult cognitive function
“Rare Genetic Variants Impact Muscle Strength”, Huang et al 2022
“Genetic Prevalence and Clinical Relevance of Canine Mendelian Disease Variants in over One Million Dogs”, Donner et al 2022
“The Female Protective Effect against Autism Spectrum Disorder”, Wigdor et al 2022
The female protective effect against autism spectrum disorder
“Using Genomics to Fight Extinction: Quantifying Fitness of Wild Organisms from Genomic Data Alone Is a Challenging Frontier”, Grueber & Sunnucks 2022
“The Critically Endangered Vaquita Is Not Doomed to Extinction by Inbreeding Depression”, Robinson et al 2022
The critically endangered vaquita is not doomed to extinction by inbreeding depression
“Polygenic Risk Score As a Possible Tool for Identifying Familial Monogenic Causes of Complex Diseases”, Lu et al 2022
“The Contributions of Rare Inherited and Polygenic Risk to ASD in Multiplex Families”, Chang et al 2022
The Contributions of Rare Inherited and Polygenic Risk to ASD in Multiplex Families
“Exome-Wide Screening Identifies Novel Rare Risk Variants for Major Depression Disorder”, Cheng et al 2022b
Exome-wide screening identifies novel rare risk variants for major depression disorder
“Integrating Whole-Genome Sequencing With Multi-Omic Data Reveals the Impact of Structural Variants on Gene Regulation in the Human Brain”, Vialle et al 2022
“Characterization of Arabian Peninsula Whole Exomes: Exploring High Inbreeding Features”, Ferreira et al 2022
Characterization of Arabian Peninsula whole exomes: exploring high inbreeding features
“Genome-Wide Analyses of ADHD Identify 27 Risk Loci, Refine the Genetic Architecture and Implicate Several Cognitive Domains”, Demontis et al 2022
“Genetic Risk Factors Have a Substantial Impact on Healthy Life Years”, Jukarainen et al 2022
Genetic risk factors have a substantial impact on healthy life years
“From Variant to Function in Human Disease Genetics”, Lappalainen & MacArthur 2022
“Life Histories of Myeloproliferative Neoplasms Inferred from Phylogenies”, Williams et al 2022
Life histories of myeloproliferative neoplasms inferred from phylogenies
“Ultra-Rapid Nanopore Genome Sequencing in a Critical Care Setting”, Gorzynski et al 2022
Ultra-Rapid Nanopore Genome Sequencing in a Critical Care Setting
“Rare Genetic Variants Correlate With Better Processing Speed”, Song et al 2022
Rare Genetic Variants Correlate with Better Processing Speed
“Rare Schizophrenia Risk Variant Burden Is Conserved in Diverse Human Populations”, Liu et al 2022
Rare schizophrenia risk variant burden is conserved in diverse human populations
“Schizophrenia-Associated Somatic Copy Number Variants from 12,834 Cases Reveal Contribution to Risk and Recurrent, Isoform-Specific NRXN1 Disruptions”, Maury et al 2022
“Rare Genetic Variants in Genes and Loci Linked to Dominant Monogenic Developmental Disorders Cause Milder Related Phenotypes in the General Population”, Kingdom et al 2022
“Dominant Cone Rod Dystrophy, Previously Assigned to a Missense Variant in RIMS1, Is Fully Explained by Co-Inheritance of a Dominant Allele of PROM1”, Martin-Gutierrez et al 2022
“The Origins and Functional Effects of Postzygotic Mutations throughout the Human Lifespan”, Rockweiler et al 2021
The origins and functional effects of postzygotic mutations throughout the human lifespan
“High-Impact Rare Genetic Variants in Severe Schizophrenia”, Zoghbi et al 2021
“Familial Risk and Heritability of Intellectual Disability: a Population-Based Cohort Study in Sweden”, Lichtenstein et al 2021
Familial risk and heritability of intellectual disability: a population-based cohort study in Sweden
“CONGA: Copy Number Variation Genotyping in Ancient Genomes and Low-Coverage Sequencing Data”, Soylev et al 2021
CONGA: Copy number variation genotyping in ancient genomes and low-coverage sequencing data
“A Spectrum of Recessiveness among Mendelian Disease Variants in UK Biobank”, Barton et al 2021
A spectrum of recessiveness among Mendelian disease variants in UK Biobank
“Fine-Scale Population Structure and Demographic History of British Pakistanis”, Arciero et al 2021
Fine-scale population structure and demographic history of British Pakistanis
“The Effect of Inbreeding, Body Size and Morphology on Health in Dog Breeds”, Bannasch et al 2021
The effect of inbreeding, body size and morphology on health in dog breeds
“Deletion of Loss-Of-Function-Intolerant Genes and Risk of 5 Psychiatric Disorders”, Wainberg et al 2021
Deletion of Loss-of-Function-Intolerant Genes and Risk of 5 Psychiatric Disorders
“Exploring the Relationships between Autozygosity, Educational Attainment, and Cognitive Ability in a Contemporary, Trans-Ancestral American Sample”, Colbert et al 2021
“Deep Learning Enables Genetic Analysis of the Human Thoracic Aorta”, Pirruccello et al 2021
Deep learning enables genetic analysis of the human thoracic aorta
“Comparing Copy Number Variations in a Danish Case Cohort of Individuals With Psychiatric Disorders”, Sánchez et al 2021
Comparing Copy Number Variations in a Danish Case Cohort of Individuals With Psychiatric Disorders
“The Sequences of 150,119 Genomes in the UK Biobank”, Halldorsson et al 2021
“The Impact of Rare Germline Variants on Human Somatic Mutation Processes”, Vali-Pour et al 2021
The impact of rare germline variants on human somatic mutation processes
“100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report”, Investigators 2021
100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care—Preliminary Report
“Influences of Rare Copy Number Variation on Human Complex Traits”, Hujoel et al 2021
Influences of rare copy number variation on human complex traits
“Rare Variant Aggregation in 148,508 Exomes Identifies Genes Associated With Proxy Alzheimer’s Disease”, Wightman et al 2021
“Integrating de Novo and Inherited Variants in over 42,607 Autism Cases Identifies Mutations in New Moderate Risk Genes”, Zhou et al 2021
“How Rare and Common Risk Variation Jointly Affect Liability for Autism Spectrum Disorder”, Klei et al 2021
How rare and common risk variation jointly affect liability for autism spectrum disorder
“A General Framework for Identifying Rare Variant Combinations in Complex Disorders”, Pounraja & Girirajan 2021
A general framework for identifying rare variant combinations in complex disorders
“Extreme Purifying Selection against Point Mutations in the Human Genome”, Dukler et al 2021
Extreme purifying selection against point mutations in the human genome
“Rates of Contributory de Novo Mutation in High and Low-Risk Autism Families”, Yoon et al 2021
Rates of contributory de novo mutation in high and low-risk autism families
“Partitioning Gene-Level Contributions to Complex-Trait Heritability by Allele Frequency Identifies Disease-Relevant Genes”, Burch et al 2021
“Differences in the Genetic Architecture of Common and Rare Variants in Childhood, Persistent and Late-Diagnosed Attention Deficit Hyperactivity Disorder”, Rajagopal et al 2021
“Genetic Correlates of Phenotypic Heterogeneity in Autism”, Warrier et al 2021
“Sequencing of 640,000 Exomes Identifies GPR75 Variants Associated With Protection from Obesity”, Akbari et al 2021
Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity
“Finding Genes That Control Body Weight: DNA Exome Sequencing at Scale Reveals Unknown Human Biology of Adiposity”, Yeo & O’Rahilly 2021
“Genomic Partitioning of Inbreeding Depression in Humans”, Yengo et al 2021
“Exome Sequencing in Obsessive-Compulsive Disorder Reveals a Burden of Rare Damaging Coding Variants”, Halvorsen et al 2021
Exome sequencing in obsessive-compulsive disorder reveals a burden of rare damaging coding variants
“Recovery of Trait Heritability from Whole Genome Sequence Data”, Wainschtein et al 2021
Recovery of trait heritability from whole genome sequence data
“Ultra-Rare, Rare, and Common Genetic Variant Analysis Converge to Implicate Negative Selection and Neuronal Processes in the Aetiology of Schizophrenia”, Akingbuwa et al 2021
“Lack of Transgenerational Effects of Ionizing Radiation Exposure from the Chernobyl Accident”, Yeager et al 2021
Lack of transgenerational effects of ionizing radiation exposure from the Chernobyl accident
“A Phenotypic Spectrum of Autism Is Attributable to the Combined Effects of Rare Variants, Polygenic Risk and Sex”, Antaki et al 2021
“Structural Variants in Chinese Population and Their Impact on Phenotypes, Diseases and Population Adaptation”, Wu et al 2021
“Polygenic Burden Has Broader Impact on Health, Cognition, and Socioeconomic Outcomes Than Most Rare and High-Risk Copy Number Variants”, Saarentaus et al 2021
“A Cross-Disorder Dosage Sensitivity Map of the Human Genome”, Collins et al 2021
“Protein-Coding Repeat Polymorphisms Strongly Shape Diverse Human Phenotypes”, Mukamel et al 2021
Protein-coding repeat polymorphisms strongly shape diverse human phenotypes
“The Landscape of Somatic Mutation in Cerebral Cortex of Autistic and Neurotypical Individuals Revealed by Ultra-Deep Whole-Genome Sequencing”, Rodin et al 2021
“Large Mosaic Copy Number Variations Confer Autism Risk”, Sherman et al 2021b
“Long Tracks of Homozygosity Predict the Severity of Alcohol Use Disorders in an American Indian Population”, Peng & Ehlers 2021
“Exome Sequencing and Analysis of 454,787 UK Biobank Participants”, Backman et al 2021
Exome sequencing and analysis of 454,787 UK Biobank participants
“Long Read Sequencing of 3,622 Icelanders Provides Insight into the Role of Structural Variants in Human Diseases and Other Traits”, Beyter et al 2020
“A Broad Exome Study of the Genetic Architecture of Asthma Reveals Novel Patient Subgroups”, Cameron-Christie et al 2020
A broad exome study of the genetic architecture of asthma reveals novel patient subgroups
“Rare Genetic Variation Underlying Human Diseases and Traits: Results from 200,000 Individuals in the UK Biobank”, Jurgens et al 2020
“Discovery of Rare Variants Associated With Blood Pressure Regulation through Meta-Analysis of 1.3 Million Individuals”, Surendran et al 2020
“Exome Sequencing and Characterization of 49,960 Individuals in the UK Biobank”, Hout et al 2020
Exome sequencing and characterization of 49,960 individuals in the UK Biobank
“Mutations in Metabotropic Glutamate Receptor 1 Contribute to Natural Short Sleep Trait”, Shi et al 2020
Mutations in Metabotropic Glutamate Receptor 1 Contribute to Natural Short Sleep Trait
“Exome Sequencing Identifies Rare Coding Variants in 10 Genes Which Confer Substantial Risk for Schizophrenia”, Singh et al 2020
“Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia”, Consortium et al 2020
Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia
“Effect Sizes of Deletions and Duplications on Autism Risk Across the Genome”, Douard et al 2020
Effect Sizes of Deletions and Duplications on Autism Risk Across the Genome
“Novel Ultra-Rare Exonic Variants Identified in a Founder Population Implicate Cadherins in Schizophrenia”, Lencz et al 2020
“Reconstructing the History of Founder Events Using Genome-Wide Patterns of Allele Sharing across Individuals”, Tournebize et al 2020
“Whole-Exome Imputation within UK Biobank Powers Rare Coding Variant Association and Fine-Mapping Analyses”, Barton et al 2020
“Exome-Wide Association Studies in General and Long-Lived Populations Identify Genetic Variants Related to Human Age”, Sin-Chan et al 2020
“Genomic Analyses Implicate Noncoding de Novo Variants in Congenital Heart Disease”, Richter et al 2020
Genomic analyses implicate noncoding de novo variants in congenital heart disease
“Genetic Ancestry Analysis on >93,000 Individuals Undergoing Expanded Carrier Screening Reveals Limitations of Ethnicity-Based Medical Guidelines”, Kaseniit et al 2020
“The Burden of Rare Protein-Truncating Genetic Variants on Human Lifespan”, Liu et al 2020
The burden of rare protein-truncating genetic variants on human lifespan
“An Integrated Polygenic and Clinical Risk Tool Enhances Coronary Artery Disease Prediction”, Aguilera et al 2020
An integrated polygenic and clinical risk tool enhances coronary artery disease prediction
“Sex-Biased Reduction in Reproductive Success Drives Selective Constraint on Human Genes”, Gardner et al 2020
Sex-biased reduction in reproductive success drives selective constraint on human genes
“Etiology of Autism Spectrum Disorders and Autistic Traits Over Time”, Taylor et al 2020
Etiology of Autism Spectrum Disorders and Autistic Traits Over Time
“Estimating the Effect-Size of Gene Dosage on Cognitive Ability across the Coding Genome”, Huguet et al 2020
Estimating the effect-size of gene dosage on cognitive ability across the coding genome
“Inbreeding and Inbreeding Depression in Linebred Beef Cattle”, Hieber 2020
Inbreeding and Inbreeding Depression in Linebred Beef Cattle
“Whole-Genome Sequencing of Rare Disease Patients in a National Healthcare System”, Ouwehand et al 2020
Whole-genome sequencing of rare disease patients in a national healthcare system
“Figure 6. The Contributions of Ultra-Rare PTVs [protein-Truncating Variants] to Schizophrenia Risk. A: Genetic Architecture of Schizophrenia. Statistically-Significant Genetic Associations for Schizophrenia from the Most Recent GWAS, CNV, and Sequencing Studies Are Displayed. The In-Sample Odds Ratio Is Plotted against the Minor Allele Frequency in the General Population. The Color of Each Dot Corresponds to the Source of the Association, and the Size of the Dot to the Odds Ratio. The Shaded Area Represented the LOESS-Smoothed Lines of the Upper and Lower Bounds of the Point Estimates...Because Schizophrenia As a Trait Is under Strong Selection^38–40^, We Expect That URVs of Large Effect to Be Frequently de Novo or of Very Recent Origin and Contribute to Risk in Only a Fraction of Diagnosed Patients.”
![Figure 6. The contributions of ultra-rare PTVs [protein-truncating variants] to schizophrenia risk. A: Genetic architecture of schizophrenia. Statistically-significant genetic associations for schizophrenia from the most recent GWAS, CNV, and sequencing studies are displayed. The in-sample odds ratio is plotted against the minor allele frequency in the general population. The color of each dot corresponds to the source of the association, and the size of the dot to the odds ratio. The shaded area represented the LOESS-smoothed lines of the upper and lower bounds of the point estimates...Because schizophrenia as a trait is under strong selection^38–40^, we expect that URVs of large effect to be frequently de novo or of very recent origin and contribute to risk in only a fraction of diagnosed patients.](/doc/genetics/heritable/rare/2020-singh-figure6a-thecontributionsofultrarareptvstoschizophreniarisk.png){kind=link}
“Extended Data Figure 2: GWAS Progress over Time. The Relationship of GWAS Associations to Sample-Size Is Shown in This Plot With Selected SCZ GWAS Meta-Analyses of the past 11 Years. The X-Axis Shows Number of Cases. The Y-Axis Shows the Number of Independent Loci Discovered With at Least One Genome-Wide Statistically-Significant Index SNP in the Discovery Meta-Analysis (eg. without Replication Data)...The Slope of ~4 Newly Discovered Loci per 1000 Cases 2013–2019 Increased to a Slope of ~6 With the Latest Sample-Size Increase.”
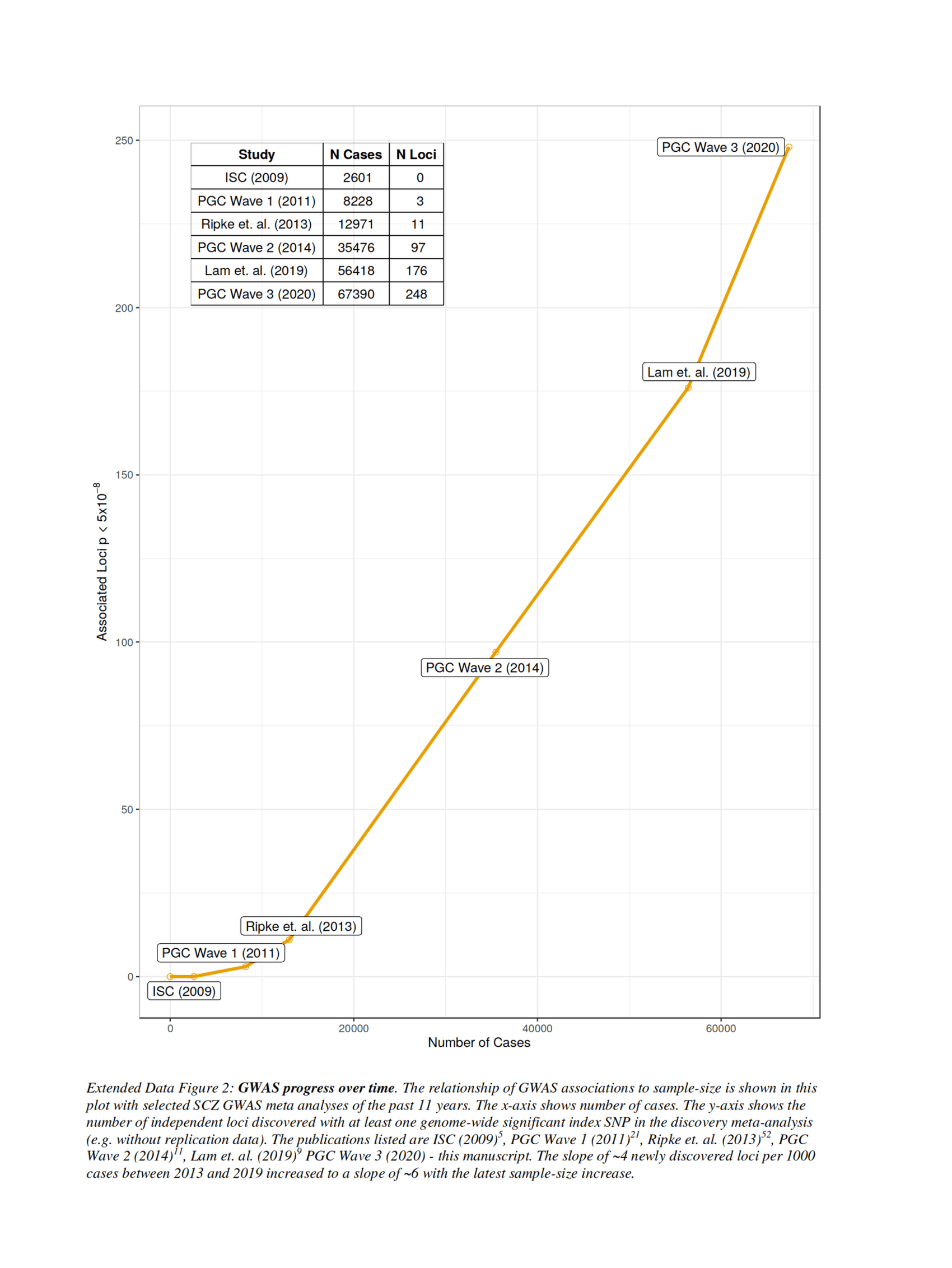{kind=link}
“Rare Genetic Variants Associated With Sudden Cardiac Death in Adults”, Khera et al 2019
Rare Genetic Variants Associated With Sudden Cardiac Death in Adults
“Mutant Neuropeptide S Receptor Reduces Sleep Duration With Preserved Memory Consolidation”, Xing et al 2019
Mutant neuropeptide S receptor reduces sleep duration with preserved memory consolidation
“Germline Burden of Rare Damaging Variants Negatively Affects Human Healthspan and Lifespan”, Shindyapina et al 2019
Germline burden of rare damaging variants negatively affects human healthspan and lifespan
“Extreme Inbreeding in a European Ancestry Sample from the Contemporary UK Population”, Yengo et al 2019
Extreme inbreeding in a European ancestry sample from the contemporary UK population
“Insights about Variation in Meiosis from 31,228 Human Sperm Genomes”, Bell et al 2019
Insights about variation in meiosis from 31,228 human sperm genomes
“The Family That Feels Almost No Pain: An Italian Clan’s Curious Insensitivity to Pain Has Piqued the Interest of Geneticists Seeking a New Understanding of How to Treat Physical Suffering”, Shaer 2019
“Symposium Review: The Genomic Architecture of Inbreeding: How Homozygosity Affects Health and Performance”, Baes et al 2019
“Crowdfunded Whole-Genome Sequencing of the Celebrity Cat Lil BUB Identifies Causal Mutations for Her Osteopetrosis and Polydactyly”, Bridavsky et al 2019
“Microdeletion in a FAAH Pseudogene Identified in a Patient With High Anandamide Concentrations and Pain Insensitivity”, Habib et al 2019
“Whole-Genome Deep-Learning Analysis Identifies Contribution of Noncoding Mutations to Autism Risk”, Zhou et al 2019b
Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk
“The Genetic Basis of Inbreeding Depression in Potato”, Zhang et al 2019
“A Rare Mutation of Β1-Adrenergic Receptor Affects Sleep/Wake Behaviors”, Shi et al 2019b
A Rare Mutation of β1-Adrenergic Receptor Affects Sleep/Wake Behaviors
“Phenome-Wide Burden of Copy-Number Variation in the UK Biobank”, Aguirre et al 2019
Phenome-wide Burden of Copy-Number Variation in the UK Biobank
“The Human-Specific BOLA2 Duplication Modifies Iron Homeostasis and Anemia Predisposition in Chromosome 16p11.2 Autism Individuals”, Giannuzzi et al 2019
“Schizophrenia Risk Conferred by Protein-Coding de Novo Mutations”, Howrigan et al 2018
Schizophrenia risk conferred by protein-coding de novo mutations
“Pfizer Terminates Domagrozumab (PF-06252616) Clinical Studies for the Treatment of Duchenne Muscular Dystrophy”, Pfizer 2018
“Quantifying the Effects of 16p11.2 Copy Number Variants on Brain Structure: A Multisite Genetic-First Study”, Martin-Brevet et al 2018
“LY2495655, an Antimyostatin Antibody, in Pancreatic Cancer: a Randomized, Phase 2 Trial”, Golan et al 2018
LY2495655, an antimyostatin antibody, in pancreatic cancer: a randomized, phase 2 trial
“Common Genetic Variants Contribute to Risk of Rare Severe Neurodevelopmental Disorders”, Niemi et al 2018
Common genetic variants contribute to risk of rare severe neurodevelopmental disorders
“Analysis of the Genetic Basis of Height in Large Jewish Nuclear Families”, Zeevi et al 2018
Analysis of the genetic basis of height in large Jewish nuclear families
“Frequency and Distribution of 152 Genetic Disease Variants in over 100,000 Mixed Breed and Purebred Dogs”, Donner et al 2018
“Relationships between Estimated Autozygosity and Complex Traits in the UK Biobank”, Johnson et al 2018
Relationships between estimated autozygosity and complex traits in the UK Biobank
“Myostatin: 20 Years Later”, Kostyunina et al 2018
“An Analytical Framework for Whole-Genome Sequence Association Studies and Its Implications for Autism Spectrum Disorder”, Werling et al 2018
“1 in 38 Individuals at Risk of a Dominant Medically Actionable Disease”, Haer-Wigman et al 2018
1 in 38 individuals at risk of a dominant medically actionable disease
“Secondary Findings from Clinical Genomic Sequencing: Prevalence, Patient Perspectives, Family History Assessment, and Health-Care Costs from a Multisite Study”, Hart et al 2018
“Medical Consequences of Pathogenic CNVs in Adults: Analysis of the UK Biobank”, Crawford et al 2018
Medical consequences of pathogenic CNVs in adults: analysis of the UK Biobank
“Measuring and Estimating the Effect Sizes of Copy Number Variants on General Intelligence in Community-Based Samples”, Huguet et al 2018
“Singleton Variants Dominate the Genetic Architecture of Human Gene Expression”, Hernandez et al 2017
Singleton Variants Dominate the Genetic Architecture of Human Gene Expression
“CNV-Association Meta-Analysis in 191,161 European Adults Reveals New Loci Associated With Anthropometric Traits”, Macé et al 2017
“Quantification of Frequency-Dependent Genetic Architectures and Action of Negative Selection in 25 UK Biobank Traits”, Schoech et al 2017
“A Genome-Wide Association Study for Extremely High Intelligence”, Zabaneh et al 2017
A genome-wide association study for extremely high intelligence
“The Surprising Implications of Familial Association in Disease Risk”, Valberg et al 2017
The surprising implications of familial association in disease risk
“Quantifying the Impact of Rare and Ultra-Rare Coding Variation across the Phenotypic Spectrum”, Ganna et al 2017
Quantifying the impact of rare and ultra-rare coding variation across the phenotypic spectrum
“Genomic Analysis of Family Data Reveals Additional Genetic Effects on Intelligence and Personality”, Hill et al 2017
Genomic analysis of family data reveals additional genetic effects on intelligence and personality
“Prevalence and Architecture of de Novo Mutations in Developmental Disorders”, McRae et al 2017
Prevalence and architecture of de novo mutations in developmental disorders
“Excess of Genomic Defects in a Woolly Mammoth on Wrangel Island”, Rogers & Slatkin 2017
Excess of genomic defects in a woolly mammoth on Wrangel island
“Parental Influence on Human Germline de Novo Mutations in 1,548 Trios from Iceland”, Jónsson et al 2017
Parental influence on human germline de novo mutations in 1,548 trios from Iceland
“Inequality in Genetic Cancer Risk Suggests Bad Genes rather than Bad Luck”, Stensrud & Valberg 2017
Inequality in genetic cancer risk suggests bad genes rather than bad luck
“Polygenic Transmission Disequilibrium Confirms That Common and Rare Variation Act Additively to Create Risk for Autism Spectrum Disorders”, Weiner et al 2016
“Trans-Ancestry Meta-Analyses Identify Rare and Common Variants Associated With Blood Pressure and Hypertension”, Surendran et al 2016
“Clinical Utility of Expanded Carrier Screening: Reproductive Behaviors of At-Risk Couples”, Ghiossi et al 2016
Clinical Utility of Expanded Carrier Screening: Reproductive Behaviors of At-Risk Couples
“Extreme Distribution of Deleterious Variation in a Historically Small and Isolated Population—Insights from the Greenlandic Inuit”, Pedersen et al 2016
“A Prospective Study of Sudden Cardiac Death among Children and Young Adults”, Bagnall et al 2016
A Prospective Study of Sudden Cardiac Death among Children and Young Adults
“Cognitive Performance Among Carriers of Pathogenic Copy Number Variants: Analysis of 152,000 UK Biobank Subjects”, Kendall et al 2016
“Family-Specific Variants and the Limits of Human Genetics”, Shirts et al 2016
“Whole-Genome Sequencing of Quartet Families With Autism Spectrum Disorder”
Whole-genome sequencing of quartet families with autism spectrum disorder
“The Relative Contribution of Common and Rare Genetic Variants to ADHD”, Martin 2015
The relative contribution of common and rare genetic variants to ADHD
“Complete Genomes Reveal Signatures of Demographic and Genetic Declines in the Woolly Mammoth”, Palkopoulou et al 2015
Complete genomes reveal signatures of demographic and genetic declines in the woolly mammoth
“Directional Dominance on Stature and Cognition in Diverse Human Populations”, Joshi et al 2015
Directional dominance on stature and cognition in diverse human populations
“Synaptic, Transcriptional and Chromatin Genes Disrupted in Autism”, Rubeis et al 2014
Synaptic, transcriptional and chromatin genes disrupted in autism
“Estimating the Inbreeding Depression on Cognitive Behavior: A Population Based Study of Child Cohort”, Fareed & Afzal 2014
Estimating the Inbreeding Depression on Cognitive Behavior: A Population Based Study of Child Cohort
“A Novel BHLHE41 Variant Is Associated With Short Sleep and Resistance to Sleep Deprivation in Humans”, Pellegrino et al 2014
A novel BHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans
“Large-Scale Genomics Unveils the Genetic Architecture of Psychiatric Disorders”, Gratten et al 2014
Large-scale genomics unveils the genetic architecture of psychiatric disorders
“The Contribution of de Novo Coding Mutations to Autism Spectrum Disorder”, Iossifov et al 2014
The contribution of de novo coding mutations to autism spectrum disorder
“The Effect of Paternal Age on Offspring Intelligence and Personality When Controlling for Paternal Trait Level”, Arslan et al 2013
“Analysis of 6,515 Exomes Reveals the Recent Origin of Most Human Protein-Coding Variants”, Fu et al 2013
Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants
“The Incidence of Leukemia, Lymphoma and Multiple Myeloma among Atomic Bomb Survivors: 1950-2001”, Hsu et al 2013
The incidence of leukemia, lymphoma and multiple myeloma among atomic bomb survivors: 1950-2001
“Range of Genetic Mutations Associated With Severe Non-Syndromic Sporadic Intellectual Disability: an Exome Sequencing Study”, Rauch et al 2012
“CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics”, Malhotra & Sebat 2012
CNVs: harbingers of a rare variant revolution in psychiatric genetics
“Heritability of Performance Deficit Accumulation during Acute Sleep Deprivation in Twins”, Kuna et al 2012
Heritability of performance deficit accumulation during acute sleep deprivation in twins
“Common Variants Show Predicted Polygenic Effects on Height in the Tails of the Distribution, Except in Extremely Short Individuals”, Chan et al 2011
“Rare Copy Number Deletions Predict Individual Variation in Intelligence”, Yeo et al 2010
Rare Copy Number Deletions Predict Individual Variation in Intelligence
“Genomic Insights into Early-Onset Obesity”, Choquet & Meyre 2010
“Population-Based Carrier Screening for Cystic Fibrosis in Victoria: The First 3 Years Experience”, Massie et al 2009
Population-based carrier screening for cystic fibrosis in Victoria: The first 3 years experience
“An Expressed fgf4 Retrogene Is Associated With Breed-Defining Chondrodysplasia in Domestic Dogs.”, Parker et al 2009
An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs.
“The Transcriptional Repressor DEC2 Regulates Sleep Length in Mammals”, He et al 2009
The transcriptional repressor DEC2 regulates sleep length in mammals
“The VNTR 2 Repeat in MAOA and Delinquent Behavior in Adolescence and Young Adulthood: Associations and MAOA Promoter Activity”, Guo et al 2008
“Language and Communicative Development in Williams Syndrome”, Mervis & Becerra 2007
“Genetic Enhancement of Cognition in a Kindred With Cone-Rod Dystrophy due to RIMS1 Mutation”, Sisodiya et al 2007
Genetic enhancement of cognition in a kindred with cone-rod dystrophy due to RIMS1 mutation
“Self-Management of Fatal Familial Insomnia. Part 2: Case Report”, Schenkein & Montagna 2006
Self-management of fatal familial insomnia. Part 2: case report
“Myostatin Mutation Associated With Gross Muscle Hypertrophy in a Child”, Schuelke et al 2004
Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child
“Influence of Five Years of Antenatal Screening on the Paediatric Cystic Fibrosis Population in One Region”, Cunningham & Marshall 1998
“When Kim Goodsell Discovered That She Had Two Extremely Rare Genetic Diseases, She Taught Herself Genetics to Help Find out Why.”
“Natural History of Ashkenazi Intelligence”
“The Sports Gene: Inside the Science of Extraordinary Athletic Performance”
The Sports Gene: Inside the Science of Extraordinary Athletic Performance
“Fathers Bequeath More Mutations As They Age: Genome Study May Explain Links between Paternal Age and Conditions such as Autism”
“Monkeys Genetically Modified to Show Autism Symptoms: But It Is Unclear How Well the Results Match the Condition in Humans”
“A Genome-Wide Analysis of Putative Functional and Exonic Variation Associated With Extremely High Intelligence”
“A Gene That Makes You Need Less Sleep?”
“What's Behind Many Mystery Ailments? Genetic Mutations, Study Finds”
What's Behind Many Mystery Ailments? Genetic Mutations, Study Finds
“Thinking Positively: The Genetics of High Intelligence”
“Why Do Humans Still Have a Gene That Increases the Risk of Alzheimer’s?”
Why Do Humans Still Have a Gene That Increases the Risk of Alzheimer’s?
Sort By Magic
Annotations sorted by machine learning into inferred 'tags'. This provides an alternative way to browse: instead of by date order, one can browse in topic order. The 'sorted' list has been automatically clustered into multiple sections & auto-labeled for easier browsing.
Beginning with the newest annotation, it uses the embedding of each annotation to attempt to create a list of nearest-neighbor annotations, creating a progression of topics. For more details, see the link.
disease-genetics
rare-diseases
schizophrenia-genetics
genomics
Wikipedia
Miscellaneous
-
/doc/genetics/heritable/rare/2021-saarentaus-figure3-healthimpactcnvspgsfinnish.png: -
https://ajp.psychiatryonline.org/doi/abs/10.1176/appi.ajp.2020.19080834 -
https://davidepstein.substack.com/p/sudden-cardiac-death-in-athletes -
https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-021-00855-5 -
https://infoproc.blogspot.com/2014/02/hints-of-genomic-dark-matter-rare.html -
https://story.californiasunday.com/superhero-gene-euan-ashley-stanford/: -
https://www.newyorker.com/magazine/2013/09/09/man-and-superman -
https://www.newyorker.com/magazine/2014/07/21/one-of-a-kind-2 -
https://www.newyorker.com/science/maria-konnikova/practice-doesnt-make-perfect -
https://www.nytimes.com/2017/07/14/opinion/sunday/alzheimers-cure-south-america.html -
https://www.outsideonline.com/culture/books-media/how-athletes-get-great/: -
https://www.propublica.org/article/muscular-dystrophy-patient-olympic-medalist-same-genetic-mutation: -
https://www.quantamagazine.org/animal-mutation-rates-reveal-traits-that-speed-evolution-20230405/ -
https://www.quantamagazine.org/how-pools-of-genetic-diversity-affect-a-species-fate-20230425/ -
https://www.reddit.com/r/science/comments/2l9dpi/two_new_studies_published_in_nature_provide/: -
https://www.spring.org.uk/2014/11/autism-new-studies-identify-dozens-more-associated-genes.php -
https://www.theatlantic.com/health/archive/2024/03/dna-tests-incest/677791/ -
https://www.wired.com/story/sleep-no-more-crusade-genetic-killer/
{kind=link}
{kind=link}
{kind=link}
Link Bibliography
-
https://www.medrxiv.org/content/10.1101/2024.04.03.24305256.full: “Widespread Recessive Effects on Common Diseases in a Cohort of 44,000 British Pakistanis and Bangladeshis With High Autozygosity”, -
https://www.nytimes.com/2023/07/20/magazine/family-genetics-frontotemporal-dementia.html: “The Vanishing Family: They All Have a 50-50 Chance of Inheriting a Cruel Genetic Mutation—Which Means Disappearing into Dementia in Middle Age. This Is the Story of What It’s like to Live With Those Odds”, Robert Kolker -
https://www.nature.com/articles/s41591-022-02046-0: “Rare and Common Genetic Determinants of Metabolic Individuality and Their Effects on Human Health”, -
2022-grueber.pdf: “Using Genomics to Fight Extinction: Quantifying Fitness of Wild Organisms from Genomic Data Alone Is a Challenging Frontier”, Catherine E. Grueber, Paul Sunnucks -
2022-cheng-2.pdf: “Exome-Wide Screening Identifies Novel Rare Risk Variants for Major Depression Disorder”, -
2022-williams.pdf: “Life Histories of Myeloproliferative Neoplasms Inferred from Phylogenies”, -
2022-gorzynski.pdf: “Ultra-Rapid Nanopore Genome Sequencing in a Critical Care Setting”, -
2021-yeo.pdf: “Finding Genes That Control Body Weight: DNA Exome Sequencing at Scale Reveals Unknown Human Biology of Adiposity”, Giles S. H. Yeo, Stephen O’Rahilly